Assessing the Diversity and Distribution of Apicomplexans in Host and Free-Living Environments Using High-Throughput Amplicon Da
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Basal Body Structure and Composition in the Apicomplexans Toxoplasma and Plasmodium Maria E
Francia et al. Cilia (2016) 5:3 DOI 10.1186/s13630-016-0025-5 Cilia REVIEW Open Access Basal body structure and composition in the apicomplexans Toxoplasma and Plasmodium Maria E. Francia1* , Jean‑Francois Dubremetz2 and Naomi S. Morrissette3 Abstract The phylum Apicomplexa encompasses numerous important human and animal disease-causing parasites, includ‑ ing the Plasmodium species, and Toxoplasma gondii, causative agents of malaria and toxoplasmosis, respectively. Apicomplexans proliferate by asexual replication and can also undergo sexual recombination. Most life cycle stages of the parasite lack flagella; these structures only appear on male gametes. Although male gametes (microgametes) assemble a typical 9 2 axoneme, the structure of the templating basal body is poorly defined. Moreover, the rela‑ tionship between asexual+ stage centrioles and microgamete basal bodies remains unclear. While asexual stages of Plasmodium lack defined centriole structures, the asexual stages of Toxoplasma and closely related coccidian api‑ complexans contain centrioles that consist of nine singlet microtubules and a central tubule. There are relatively few ultra-structural images of Toxoplasma microgametes, which only develop in cat intestinal epithelium. Only a subset of these include sections through the basal body: to date, none have unambiguously captured organization of the basal body structure. Moreover, it is unclear whether this basal body is derived from pre-existing asexual stage centrioles or is synthesized de novo. Basal bodies in Plasmodium microgametes are thought to be synthesized de novo, and their assembly remains ill-defined. Apicomplexan genomes harbor genes encoding δ- and ε-tubulin homologs, potentially enabling these parasites to assemble a typical triplet basal body structure. -

Implications of Habitat Restoration for Bumble Bee Population Dynamics, Foraging Ecology, and Epidemiology
IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen A DISSERTATION Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Entomology—Doctor of Philosophy Ecology, Evolutionary Biology and Behavior—Dual Major 2018 ABSTRACT IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen Many insects provide valuable ecosystem services, including those that support our food supply. Beneficial insects such as pollinators fulfill part of this role by contributing to approximately one third of the global food crop production. Over the past few decades, pollinators have faced declining populations due to a variety of factors such as agricultural intensification, lack of floral and nesting resources, and disease. One method used in agricultural settings to help sustain pollinator populations is designating unfarmed habitat such as ditches and field margins for habitat enhancement in the form of hedgerows and wildflower strips. These floristically rich areas can be tailored to bloom both before and after crop bloom to help sustain pollinators during the time when crops are not in bloom. In turn, bee populations can benefit from the consistent availability of resources in these areas of habitat enhancement. This dissertation explores how habitat enhancement affects nesting density of a common wild pollinator, Bombus impatiens . Further, this research also aims to determine how foraging preferences change and how bumble bee disease transmission and prevalence respond to habitat enhancement. Research was conducted at 15 commercial highbush blueberry ( Vaccinium corymbosum ) fields in southwest Michigan containing either no restoration, a newly planted restoration, or a mature (5-8 year old) restoration in the field margin from 2015 to 2017. -

Identification of a Novel Fused Gene Family Implicates Convergent
Chen et al. BMC Genomics (2018) 19:306 https://doi.org/10.1186/s12864-018-4685-y RESEARCH ARTICLE Open Access Identification of a novel fused gene family implicates convergent evolution in eukaryotic calcium signaling Fei Chen1,2,3, Liangsheng Zhang1, Zhenguo Lin4 and Zong-Ming Max Cheng2,3* Abstract Background: Both calcium signals and protein phosphorylation responses are universal signals in eukaryotic cell signaling. Currently three pathways have been characterized in different eukaryotes converting the Ca2+ signals to the protein phosphorylation responses. All these pathways have based mostly on studies in plants and animals. Results: Based on the exploration of genomes and transcriptomes from all the six eukaryotic supergroups, we report here in Metakinetoplastina protists a novel gene family. This family, with a proposed name SCAMK,comprisesSnRK3 fused calmodulin-like III kinase genes and was likely evolved through the insertion of a calmodulin-like3 gene into an SnRK3 gene by unequal crossover of homologous chromosomes in meiosis cell. Its origin dated back to the time intersection at least 450 million-year-ago when Excavata parasites, Vertebrata hosts, and Insecta vectors evolved. We also analyzed SCAMK’s unique expression pattern and structure, and proposed it as one of the leading calcium signal conversion pathways in Excavata parasite. These characters made SCAMK gene as a potential drug target for treating human African trypanosomiasis. Conclusions: This report identified a novel gene fusion and dated its precise fusion time -

Prevalence of Cryptosporidium Spp. \(Eucoccidiorida
Article available at http://www.parasite-journal.org or http://dx.doi.org/10.1051/parasite/2007144335 PREVALENCE OF CRYPTOSPORIDIUM SPP. (EUCOCCIDIORIDA: CRYPTOSPORIIDAE) IN SEVEN SPECIES OF FARM ANIMALS IN TUNISIA SOLTANE R.*, GUYOT K.**, DEI-CAS E.** & AYADI A.* Summary: Résumé : PRÉVALENCE DE CRYPTOSPORIDIUM SPP. (EUCOCCIDIORIDA : CRYPTOSPORIIDAE) CHEZ SEPT ESPÈCES D’ANIMAUX DE FERME EN TUNISIE 1,001 faecal samples were obtained from 89 sheep (lambs and adult), 184 goats, 190 horses, 178 rabbits, 110 camels, 1001 prélèvements fécaux ont été obtenus à partir de 89 moutons, 200 broiler chicken and 50 turkeys housed in farms from different 184 chèvres, 190 chevaux, 178 lapins, 110 chameaux, localities in Tunisia. All samples were analysed for 200 poulets et 50 dindes élevés dans des fermes de différentes Cryptosporidium oocysts by microscopic examination of smears localités en Tunisie. Tous les prélèvements ont été analysés pour la stained by modified Ziehl Neelsen technique. The parasite was recherche de Cryptosporidium par examen microscopique des detected in ten lambs and adult sheep (11.2 %) and nine broiler frottis colorés au Ziehl Neelsen modifié. Le parasite a été détecté chicken (4.5 %). Molecular characterization, performed in four chez dix ovins (11,2 %) et neuf poulets (4,5 %). La caractérisation animals, identified C. bovis in three lambs and C. meleagridis in moléculaire réalisée pour quatre isolats a identifié C. bovis chez one broiler chicken. This work is the first report on trois agneaux et C. meleagridis chez un poulet. Ce travail est le Cryptosporidium in farm animals in Tunisia. premier rapport sur Cryptosporidium chez des animaux de ferme en Tunisie. -

Journal of Parasitology
Journal of Parasitology Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes --Manuscript Draft-- Manuscript Number: 17-111R1 Full Title: Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes Short Title: Eimeria taggarti n. sp. in Prostate of Antechinus flavipes Article Type: Regular Article Corresponding Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc University of Melbourne Melbourne, Victoria AUSTRALIA Corresponding Author Secondary Information: Corresponding Author's Institution: University of Melbourne Corresponding Author's Secondary Institution: First Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc First Author Secondary Information: Order of Authors: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc Joanne Maree Devlin, BVSc(Hons), MVPHMgt, PhD Liliana Tatarczuch David Augustine Taggart David J Schultz Jenny A Charles Ian Beveridge Order of Authors Secondary Information: Abstract: A novel coccidian species was discovered in the prostate of an Antechinus flavipes (yellow-footed antechinus) in South Australia, during the period of post-mating male antechinus immunosuppression and mortality. This novel coccidian is unusual because it develops extra-intestinally and sporulates endogenously within the prostate gland of its mammalian host. Histological examination of prostatic tissue revealed dense aggregations of spherical and thin-walled tetrasporocystic, dizoic sporulated coccidian oocysts within tubular lumina, with unsporulated oocysts and gamogonic stages within the cytoplasm of glandular epithelial cells. This coccidian was observed occurring concurrently with dasyurid herpesvirus 1 infection of the antechinus' prostate. Eimeria- specific 18S small subunit ribosomal DNA PCR amplification was used to obtain a partial 18S rDNA nucleotide sequence from the antechinus coccidian. -

Culture of Exoerythrocytic Forms in Vitro
Advances in PARASITOLOGY VOLUME 27 Editorial Board W. H. R. Lumsden University of Dundee Animal Services Unit, Ninewells Hospital and Medical School, P.O. Box 120, Dundee DDI 9SY, UK P. Wenk Tropenmedizinisches Institut, Universitat Tubingen, D7400 Tubingen 1, Wilhelmstrasse 3 1, Federal Republic of Germany C. Bryant Department of Zoology, Australian National University, G.P.O. Box 4, Canberra, A.C.T. 2600, Australia E. J. L. Soulsby Department of Clinical Veterinary Medicine, University of Cambridge, Madingley Road, Cambridge CB3 OES, UK K. S. Warren Director for Health Sciences, The Rockefeller Foundation, 1133 Avenue of the Americas, New York, N.Y. 10036, USA J. P. Kreier Department of Microbiology, College of Biological Sciences, Ohio State University, 484 West 12th Avenue, Columbus, Ohio 43210-1292, USA M. Yokogawa Department of Parasitology, School of Medicine, Chiba University, Chiba, Japan Advances in PARASITOLOGY Edited by J. R. BAKER Cambridge, England and R. MULLER Commonwealth Institute of Parasitology St. Albans, England VOLUME 27 1988 ACADEMIC PRESS Harcourt Brace Jovanovich, Publishers London San Diego New York Boston Sydney Tokyo Toronto ACADEMIC PRESS LIMITED 24/28 Oval Road LONDON NW 1 7DX United States Edition published by ACADEMIC PRESS INC. San Diego, CA 92101 Copyright 0 1988, by ACADEMIC PRESS LIMITED All Rights Reserved No part of this book may be reproduced in any form by photostat, microfilm, or any other means, without written permission from the publishers British Library Cataloguing in Publication Data Advances in parasitology.-Vol. 27 1. Veterinary parasitology 591.2'3 SF810.A3 ISBN Cb12-031727-3 ISSN 0065-308X Typeset by Latimer Trend and Company Ltd, Plymouth, England Printed in Great Britain by Galliard (Printers) Ltd, Great Yarmouth CONTRIBUTORS TO VOLUME 27 B. -

Phylogeny and Evolution of Theileria and Babesia Parasites in Selected Wild Herbivores of Kenya
PHYLOGENY AND EVOLUTION OF THEILERIA AND BABESIA PARASITES IN SELECTED WILD HERBIVORES OF KENYA LUCY WAMUYU WAHOME MASTER OF SCIENCE (Bioinformatics and Molecular Biology) JOMO KENYATTA UNIVERSITY OF AGRICULTURE AND TECHNOLOGY 2016 Phylogeny and Evolution of Theileria and Babesia Parasites in Selected Wild Herbivores of Kenya Lucy Wamuyu Wahome A Thesis Submitted in partial fulfillment for the Degree of Master of Science in Bioinformatics and Molecular Biology in the Jomo Kenyatta University of Agriculture and Technology. 2016 DECLARATION This thesis is my original work and has not been presented for a degree in any other University. Signature………………………. Date…………………………… Wahome Lucy Wamuyu This thesis has been submitted for examination with our approval as university supervisors. Signature……………………… Date…………………………… Prof. Daniel Kariuki JKUAT, Kenya Signature……………………… Date…………………………… Dr.Sheila Ommeh JKUAT, Kenya Signature……………………… Date………………………… Dr. Francis Gakuya Kenya Wildlife Service, Kenya ii DEDICATION This work is dedicated to my beloved parents Mr. Godfrey, Mrs. Naomi and my only beloved sister Caroline for their tireless support throughout my academic journey. "I can do everything through Christ who gives me strength." iii ACKNOWLEDGEMENTS Foremost I would like to acknowledge and thank the Almighty God for blessing, protecting and guiding me throughout this period. I am most grateful to the department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology, for according me the opportunity to pursue my postgraduate studies. I would like to express my sincere gratitude to my supervisors Prof. Daniel Kariuki, Dr. Sheila Ommeh and Dr. Francis Gakuya for the useful comments, remarks and engagement through out this project. My deepest gratitude goes to Dr. -

Evidence for and Against Deformed Wing Virus Spillover from Honey Bees to Bumble Bees: a Reverse Genetic Analysis Olesya N
www.nature.com/scientificreports OPEN Evidence for and against deformed wing virus spillover from honey bees to bumble bees: a reverse genetic analysis Olesya N. Gusachenko1*, Luke Woodford1, Katharin Balbirnie‑Cumming1, Eugene V. Ryabov2 & David J. Evans1* Deformed wing virus (DWV) is a persistent pathogen of European honey bees and the major contributor to overwintering colony losses. The prevalence of DWV in honey bees has led to signifcant concerns about spillover of the virus to other pollinating species. Bumble bees are both a major group of wild and commercially‑reared pollinators. Several studies have reported pathogen spillover of DWV from honey bees to bumble bees, but evidence of a sustained viral infection characterized by virus replication and accumulation has yet to be demonstrated. Here we investigate the infectivity and transmission of DWV in bumble bees using the buf-tailed bumble bee Bombus terrestris as a model. We apply a reverse genetics approach combined with controlled laboratory conditions to detect and monitor DWV infection. A novel reverse genetics system for three representative DWV variants, including the two master variants of DWV—type A and B—was used. Our results directly confrm DWV replication in bumble bees but also demonstrate striking resistance to infection by certain transmission routes. Bumble bees may support DWV replication but it is not clear how infection could occur under natural environmental conditions. Deformed wing virus (DWV) is a widely established pathogen of the European honey bee, Apis mellifera. In synergistic action with its vector—the parasitic mite Varroa destructor—it has had a devastating impact on the health of honey bee colonies globally1,2. -
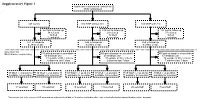
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines. -

NEW CLASSIFICATION for Toxoplasma Gondii
LETTER TO THE EDITOR NEW CLASSIFICATION FOR Toxoplasma gondii Claudio Bruno Silva de Oliveira Dear Editor, I have been following the latest publications in the field of parasitology and have noticed that, despite the changes in the group that host the parasite Toxoplasma gondii that have been suggested since 2012 (Adl et al. 2012), many articles in several journals have not been updated (Liempi et al. 2014; Ning et al. 2015; Lorenzi et al. 2016). This can be explained by a certain protectionism regarding the form that has been used for several years. However, it is important to consider that this can also be due to some unfamiliarity with the current classification of eukaryotes and protozoa. Classically this protozoan is classified within the Protista kingdom, Apicomplexa phylum, Sporozoasida class, Eucoccidiorida order, Sarcocystidae family, and Toxoplasma genus (Current et al. 1990). This classification has been used for many decades, and it is well accepted by research groups in the area. Recently, however, Adl et al. (2012) seeking to standardize and organize different groups of eukaryotes, mainly protists, suggested a new classification based on phylogenetic and ultra-structural similarity. Now Toxoplasma gondii appears within a super group called SAR comprising: Stramenopiles, Alveolata, and Rhizaria. More precisely, it appears inside the Alveolata group (first group); among Alveolata it is classified as Apicomplexa (second group); among the Apicomplexa it is classified as Conoidasida (third group); among these it is classified as Coccidia (fourth group); and finally among the Coccidia it is part of Eimeriorina group (fifth group), along with other associated parasites such as Cyclospora and Neospora. -

Why the –Omic Future of Apicomplexa Should Include Gregarines Julie Boisard, Isabelle Florent
Why the –omic future of Apicomplexa should include Gregarines Julie Boisard, Isabelle Florent To cite this version: Julie Boisard, Isabelle Florent. Why the –omic future of Apicomplexa should include Gregarines. Biology of the Cell, Wiley, 2020, 10.1111/boc.202000006. hal-02553206 HAL Id: hal-02553206 https://hal.archives-ouvertes.fr/hal-02553206 Submitted on 24 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article title: Why the –omic future of Apicomplexa should include Gregarines. Names of authors: Julie BOISARD1,2 and Isabelle FLORENT1 Authors affiliations: 1. Molécules de Communication et Adaptation des Microorganismes (MCAM, UMR 7245), Département Adaptations du Vivant (AVIV), Muséum National d’Histoire Naturelle, CNRS, CP52, 57 rue Cuvier 75231 Paris Cedex 05, France. 2. Structure et instabilité des génomes (STRING UMR 7196 CNRS / INSERM U1154), Département Adaptations du vivant (AVIV), Muséum National d'Histoire Naturelle, CP 26, 57 rue Cuvier 75231 Paris Cedex 05, France. Short Title: Gregarines –omics for Apicomplexa studies -

Protistology an International Journal Vol
Protistology An International Journal Vol. 10, Number 2, 2016 ___________________________________________________________________________________ CONTENTS INTERNATIONAL SCIENTIFIC FORUM «PROTIST–2016» Yuri Mazei (Vice-Chairman) Welcome Address 2 Organizing Committee 3 Organizers and Sponsors 4 Abstracts 5 Author Index 94 Forum “PROTIST-2016” June 6–10, 2016 Moscow, Russia Website: http://onlinereg.ru/protist-2016 WELCOME ADDRESS Dear colleagues! Republic) entitled “Diplonemids – new kids on the block”. The third lecture will be given by Alexey The Forum “PROTIST–2016” aims at gathering Smirnov (Saint Petersburg State University, Russia): the researchers in all protistological fields, from “Phylogeny, diversity, and evolution of Amoebozoa: molecular biology to ecology, to stimulate cross- new findings and new problems”. Then Sandra disciplinary interactions and establish long-term Baldauf (Uppsala University, Sweden) will make a international scientific cooperation. The conference plenary presentation “The search for the eukaryote will cover a wide range of fundamental and applied root, now you see it now you don’t”, and the fifth topics in Protistology, with the major focus on plenary lecture “Protist-based methods for assessing evolution and phylogeny, taxonomy, systematics and marine water quality” will be made by Alan Warren DNA barcoding, genomics and molecular biology, (Natural History Museum, United Kingdom). cell biology, organismal biology, parasitology, diversity and biogeography, ecology of soil and There will be two symposia sponsored by ISoP: aquatic protists, bioindicators and palaeoecology. “Integrative co-evolution between mitochondria and their hosts” organized by Sergio A. Muñoz- The Forum is organized jointly by the International Gómez, Claudio H. Slamovits, and Andrew J. Society of Protistologists (ISoP), International Roger, and “Protists of Marine Sediments” orga- Society for Evolutionary Protistology (ISEP), nized by Jun Gong and Virginia Edgcomb.