Anticholinergic Drugs Toxicity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
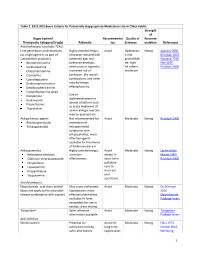
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Appendix A: Potentially Inappropriate Prescriptions (Pips) for Older People (Modified from ‘STOPP/START 2’ O’Mahony Et Al 2014)
Appendix A: Potentially Inappropriate Prescriptions (PIPs) for older people (modified from ‘STOPP/START 2’ O’Mahony et al 2014) Consider holding (or deprescribing - consult with patient): 1. Any drug prescribed without an evidence-based clinical indication 2. Any drug prescribed beyond the recommended duration, where well-defined 3. Any duplicate drug class (optimise monotherapy) Avoid hazardous combinations e.g.: 1. The Triple Whammy: NSAID + ACE/ARB + diuretic in all ≥ 65 year olds (NHS Scotland 2015) 2. Sick Day Rules drugs: Metformin or ACEi/ARB or a diuretic or NSAID in ≥ 65 year olds presenting with dehydration and/or acute kidney injury (AKI) (NHS Scotland 2015) 3. Anticholinergic Burden (ACB): Any additional medicine with anticholinergic properties when already on an Anticholinergic/antimuscarinic (listed overleaf) in > 65 year olds (risk of falls, increased anticholinergic toxicity: confusion, agitation, acute glaucoma, urinary retention, constipation). The following are known to contribute to the ACB: Amantadine Antidepressants, tricyclic: Amitriptyline, Clomipramine, Dosulepin, Doxepin, Imipramine, Nortriptyline, Trimipramine and SSRIs: Fluoxetine, Paroxetine Antihistamines, first generation (sedating): Clemastine, Chlorphenamine, Cyproheptadine, Diphenhydramine/-hydrinate, Hydroxyzine, Promethazine; also Cetirizine, Loratidine Antipsychotics: especially Clozapine, Fluphenazine, Haloperidol, Olanzepine, and phenothiazines e.g. Prochlorperazine, Trifluoperazine Baclofen Carbamazepine Disopyramide Loperamide Oxcarbazepine Pethidine -

Ketamine As a General Anesthesia Adjunct Michaela Wilcox, DNP
Ketamine as a General Anesthesia Adjunct Michaela Wilcox, DNP (c), RN Dept. of Nurse Anesthesia, Moffett & Sanders School of Nursing, Samford University Structured Abstract Background Ketamine is a unique anesthetic agent with both anesthetic and analgesic properties. Ketamine is primarily a N-Methyl-D-Aspartic acid (NMDA) antagonist that also works on glutamine, nicotinic, muscarinic, monoaminergic and opioid receptors. The uses of ketamine are numerous and include treatment of chronic pain, acute pain, depression, as a multimodal pain adjunct, opioid-sparing agent and as an adjunct in enhanced recovery after surgery (ERAS) protocols. A 49-year-old female presented for a lumbar lateral interbody fusion extreme, anterior lumbar spine arthrodesis L3/4 with revision and instrumentation. Based on the surgical need for both sensory and motor nerve monitoring intraoperatively, the anesthetic plan included minimal sevoflurane, succinylcholine for induction and a propofol intravenous (IV) infusion with boluses of ketamine to maintain unconsciousness. A bispectral index (BIS) monitor (Aspect Medical Systems, Natick, MA) was placed on the patient’s forehead prior to induction and utilized intraoperatively to maintain a goal of 40-60. General anesthesia was achieved with fentanyl 100 mcg, lidocaine 100 mg, propofol 200 mg, ketamine 50 mg and succinylcholine 160 mg IV. Laryngoscopy was successfully performed using a McGrath MAC video laryngoscope (Medtronic, Minneapolis, MN). Following induction, a propofol IV infusion was initiated and sevoflurane maintained at 0.3 minimum alveolar concentration. Ketamine 10 mg IV was administered every hour. Upon conclusion of the procedure, the patient was extubated and transported to postoperative recovery unit, where she had no complaints of nausea, vomiting or pain. -

(Antimuscarinic) Drugs?
© July - August 2018 How well do you know your anticholinergic (antimuscarinic) drugs? nticholinergic drugs, prescribed for a variety of clini- Acal conditions, are amongst the most frequently used prescription drugs in BC (Table 1). Also referred to as “an- timuscarinics,” such drugs specifically block muscarinic receptors for acetylcholine (ACh).1 Muscarinic ACh recep- tors are important in the parasympathetic nervous system that governs heart rate, exocrine glands, smooth muscles, clude drugs whose active metabolites are potent- as well as brain function. In contrast, nicotinic ACh recep- ly antimuscarinic,5 or which often cause typical tors stimulate contraction of striated muscles. This Letter is AC adverse effects such as dry mouth or urinary intended to remind clinicians of commonly used drugs that retention.6 People taking antihistamines, antide- have anticholinergic (AC), or technically, antimuscarinic pressants, antipsychotics, opioids, antimuscarinic properties, and of their potential adverse effects. inhalers, or many other drugs need to know that Beneficial and harmful effects of anticholinergic drugs have blockade of ACh receptors can cause bothersome been known for centuries. In Homer’s Odyssey, the nymph or even dangerous adverse effects (Table 3). pharmacologist Circe utilized central effects of atropinics Subtle and not-so-subtle toxicity in the common plant jimson weed (Datura stramonium) to cause delusions in the crew of Odysseus. Believing they Students often learn the adverse effects of anticho- had been turned into pigs, they could be herded.2 linergics from a mnemonic, e.g.: “Blind as a bat, Sometimes a drug is recommended specifically for its an- mad as a hatter, red as a beet, hot as a hare, dry as ticholinergic potency. -

Reference List of Drugs with Potential Anticholinergic Effects 1, 2, 3, 4, 5
ANTICHOLINERGICS: Reference List of Drugs with Potential Anticholinergic Effects 1, 2, 3, 4, 5 J Bareham BSP © www.RxFiles.ca Aug 2021 WHENEVER POSSIBLE, AVOID DRUGS WITH MODERATE TO HIGH ANTICHOLINERGIC ACTIVITY IN OLDER ADULTS (>65 YEARS OF AGE) Low Anticholinergic Activity; Moderate/High Anticholinergic Activity -B in combo Beers Antibiotics Antiparkinsonian Cardiovascular Agents Immunosuppressants ampicillin *ALL AVAILABLE AS amantadine SYMMETREL atenolol TENORMIN azaTHIOprine IMURAN cefOXitin GENERIC benztropine mesylate COGENTIN captopril CAPOTEN cyclosporine NEORAL clindamycin bromocriptine PARLODEL chlorthalidone GENERIC ONLY hydrocortisone CORTEF gentamicin (Oint & Sol’n NIHB covered) carbidopa/levodopa SINEMET digoxin LANOXIN, TOLOXIN methylprednisolone MEDROL piperacillin entacapone COMTAN dilTIAZem CARDIZEM, TIAZAC prednisone WINPRED dipyridamole PERSANTINE, ethopropazine PARSITAN vancomycin phenelzine NARDIL AGGRENOX disopyramide RYTHMODAN Muscle Relaxants pramipexole MIRAPEX Antidepressants baclofen LIORESAL ( on intrathecal only) procyclidine KEMADRIN furosemide LASIX amitriptyline ELAVIL cyclobenzaprine FLEXERIL selegiline ELDEPRYL hydrALAZINE APRESOLINE clomiPRAMINE ANAFRANIL isosorbide ISORDIL methocarbamol ROBAXIN OTC trihexyphenidyl ARTANE desipramine NORPRAMIN metoprolol LOPRESOR orphenadrine NORFLEX OTC doxepin >6mg SINEQUAN Antipsychotics NIFEdipine ADALAT tiZANidine ZANAFLEX A imipramine TOFRANIL quiNIDine GENERIC ONLY C ARIPiprazole ABILIFY & MAINTENA -

Anesthesia and Analgesia in Laboratory Animals
GUIDELINES ON ANESTHESIA AND ANALGESIA IN LABORATORY ANIMALS University of South Florida provides the following guidelines for use by IACUC-certified faculty and staff. CONTENTS PAGE A. Background……………………………………………………….…………………………… 1 B. Definitions....……………………………………………………..…………………………….. 2 C. General Considerations……………………………………….,…………………………….. 3 D. Controlled Substances……………………………………….……………………………… 3 E. Pre-Anesthetic Treatments………………………………….………………………………. 4 F. General Anesthetics………………………………………….………………………………. 4 G. Neuromuscular Blocking Agents………………………….……………………………….. 5 H. Monitoring Anesthesia…………………………………….…………………………………. 6 I. Analgesics……………………………………………………………………………………… 7 J. Comments regarding Anesthetics and Analgesics……………………………………... 7 REFERENCE TABLES PAGE I. Signs of Pain and Distress in Laboratory Animals………………………………………… 10 II. Commonly Used Anesthetics and Analgesics for Mice….………..…...….………...…… 11 III. Commonly Used Anesthetics and Analgesics for Rats……………………………...…… 12 IV. Commonly Used Anesthetics and Analgesics for Gerbils……….……………..…….. 13 V. Commonly Used Anesthetics and Analgesics for Hamsters…….……………..……. 14 VI. Commonly Used Anesthetics and Analgesics for Guinea Pigs….…………….….……. 15 VII. Commonly Used Anesthetics and Analgesics for Rabbits.……...…………….………… 16 VIII. Commonly Used Anesthetics and Analgesics for Dogs.…………………….…………… 17 IX. Commonly Used Anesthetics and Analgesics for Cats.……………………..…………… 18 X. Commonly Used Anesthetics and Analgesics for Pigs ..……………..….………………..19 XI. Commonly Used Anesthetics and Analgesics -

Metered-Dose Inhalers (Mdis): Anti-Cholinergic Drugs
Texas Vendor Drug Program Drug Use Criteria: Aerosolized Agents - Metered-Dose Inhalers (MDIs): Anti-Cholinergic Drugs Publication History 1. Developed January 1995. 2. Revised April 2021; March 2019; March 2017; November 2015; March 2014; August 2012; June 2012; October 2010; January 2008; January 2003; January 2002; January 2001; March 2000; January 2000; February 1999; February 1998; February 1997; August 1995. Notes: All criteria may be applied retrospectively. The information contained is for the convenience of the public. The Texas Health and Human Services Commission is not responsible for any errors in transmission or any errors or omissions in the document. Medications listed in the tables and non-FDA approved indications included in these retrospective criteria are not indicative of Vendor Drug Program formulary coverage. Prepared by: • Drug Information Service, UT Health San Antonio. • The College of Pharmacy, The University of Texas at Austin 1 1 Dosage 1.1 Adults Ipratropium (Atrovent®), a short-acting, inhalational anticholinergic agent, is FDA- approved to manage bronchospasm associated with chronic bronchitis and emphysema, collectively known as chronic obstructive pulmonary disease (COPD). Ipratropium is considered a second-line agent in the treatment of asthma as the bronchodilatory effects seen with ipratropium are less than those seen with beta- adrenergic drugs. While not FDA approved, the Expert Panel 3 guidelines from the National Heart Lung and Blood Institute document benefit when multiple ipratropium doses are administered adjunctively with beta2-agonists in the emergency department to manage more severe acute asthma exacerbations, and the Global Initiative for Asthma (GINA) guidelines state that ipratropium may be considered an alternative bronchodilator in patients who experience adverse effects to short-acting beta2-agonists (e.g., tachycardia, arrhythmia, tremor). -

CONTRAINDICATIONS ------Pediatric Patients (486 on DETROL LA, 224 on Placebo) Is Available
HIGHLIGHTS OF PRESCRIBING INFORMATION • Controlled Narrow-Angle Glaucoma: use caution in patients being These highlights do not include all the information needed to use Detrol® treated for narrow-angle glaucoma. (5.3) LA safely and effectively. See full prescribing information for Detrol LA. • Myasthenia Gravis: use caution in patients with myasthenia gravis. (5.6) • QT Prolongation: Consider observations from the thorough QT study in ® Detrol LA (tolterodine tartrate extended release capsules) clinical decisions to prescribe DETROL LA to patients with a known For oral administration history of QT prolongation or to patients who are taking Class IA (e.g., Initial U.S. Approval: December 2000 quinidine, procainamide) or Class III (e.g., amiodarone, sotalol) antiarrhythmic medications (5.7) ----------------------------INDICATIONS AND USAGE--------------------------- Detrol LA is an antimuscarinic indicated for the treatment of overactive ------------------------------ADVERSE REACTIONS------------------------------- bladder with symptoms of urge urinary incontinence, urgency, and frequency. The most common adverse reactions (incidence >4% and >placebo) were dry (1) mouth , headache, constipation and abdominal pain. (6.1) ----------------------DOSAGE AND ADMINISTRATION----------------------- • 4 mg capsules taken orally once daily with water and swallowed To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at whole. (2.1) 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. • 2 mg capsules taken orally once daily with water and swallowed whole in the presence of: ------------------------------DRUG INTERACTIONS------------------------------- o mild to moderate hepatic impairment (Child-Pugh • Potent CYP3A4 inhibitors: Co-administration may increase systemic class A or B) (2.2) exposure to DETROL LA. Reduce DETROL LA dose to 2 mg once o severe renal impairment [Creatinine Clearance (CCr) daily. -

(Cholinergic Antagonists) (Anticholinergic ) (Cholinergic
Parasympathlytic (Cholinergic antagonists) (Anticholinergic ) (Cholinergic Blockers) A- antimuscarinic agents (Muscarinic Antagonists): " Agents with high binding affinity for muscarinic receptors but no intrinsic activity. Pharmacologic effects opposite of the muscarinic agonists. " Competitive (reversible) antagonists of ACh " Antagonistic responses include: decreased contraction of GI and urinary tract smooth muscles, dilation of pupils, reduced gastric secretion, decreased saliva secretion. A- antimuscarinic agents (Muscarinic Antagonists): 1-Atropine (belladonna alkaloid) " (Competitive inhibitors) . -bind to muscarinic receptors and prevent Ach binding. " reversible blockade of ACh at muscarinic receptors by competitive binding! -reversal effect of atropine by increasing ACh or agonist ----> decreased blockade -atropine is central & peripheral muscarinic blocker. Muscarinic receptor blockade does not interfere with transmission at autonomic ganglionic sites, the adrenal medulla, or skeletal muscle fibers. Sympathetic adrenergic functions are not affected. X X MUSCARINIC RECEPTOR BLOCKADE ALLOWS SYMPATHETIC DOMINANCE IN DUAL INNERVATED ORGANS X Atropine actions "Eye: *mydriasis *unresponsiveness to light *cycloplegia *increase IOP " GIT: reduce activity of GIT. " Urinary system: reduce hyper motility. " Cardiovascular system: at low dose bradycardia at high dose tachycardia " Secretions: reduce secretions Therapeutic uses of atropine 1-Ophthalmic: Ophthalmologic examinations. mydriatic & cycloplegic effects. 2-antispasmotic agent -
Anticholinergic Pocket Reference Card
Anticholinergic Pocket Reference Card Because so many drugs have anticholinergic properties—and many of these are contained in over-the-counter products—anticholinergics are used by many older adults, including about 1/3 of people with dementia.1,2 The elderly are more sensitive to anticholinergic adverse effects, and people with dementia have a high risk of adverse cognitive and psychiatric effects from these drugs.3,4 Adverse effects attributed to anticholinergics include sedation, confusion, delirium, constipation, urinary retention, dry mouth, dry eyes, blurred vision, photophobia, tachycardia, decreased sweating, increased body temperature, falls, and others.5 Some evidence suggests that anticholinergics contribute to behavioral disturbances and psychosis in dementia.3 The purpose of this reference card is to help clinicians reduce anticholinergic use by vulnerable elders, especially those with cognitive impairment. Tapering may be necessary to prevent withdrawal symptoms when discontinuing potent anticholinergics that have been used chronically.2 The following lists medications with known anticholinergic effects by therapeutic use. The list is not all-inclusive, but includes many commonly used anticholinergics. Clinicians might want to especially consider the risk benefit balance of tricyclic antidepressants, immediate-release oxybutynin, GI antispasmodics, and sedating antihistamines, as these drugs are not recommended for vulnerable elders if alternative treatments are available.7 Antihistamines / Allergy / Bladder Antispasmodics Cough -

The Anticholinergic Impregnation Scale: Towards The
Therapie (2017) 72, 427—437 Available online at ScienceDirect www.sciencedirect.com CLINICAL PHARMACOLOGY The anticholinergic impregnation scale: Towards the elaboration of a scale adapted to prescriptions in French psychiatric settings L’échelle d’imprégnation anticholinergique : vers l’élaboration d’une échelle adaptée aux prescriptions en milieu psychiatrique franc¸ais a,b b,c,∗ Jeanne Briet , Hervé Javelot , c d Edwige Heitzmann , Luisa Weiner , c e Catherine Lameira , Philippe D’Athis , e a,b Marie Corneloup , Jean-Louis Vailleau a Pharmacy service, CHS de La Chartreuse, 21000 Dijon, France b PIC network (Psychiatrie Information Communication), EPSM Lille-Métropole, 59487 Armentières, France c Établissement public de santé Alsace Nord, 67170 Brumath, France d Psychiatry II and Inserm unit 1114, university hospital of Strasbourg, 67000 Strasbourg, France e Service of biostatistics and medical informatics, CHU de Dijon, 21000 Dijon, France Received 6 June 2016; accepted 23 December 2016 Available online 17 February 2017 KEYWORDS Summary Anticholinergics; Purpose. — Some drugs have anticholinergic activity and can cause peripheral or central side Anticholinergic drug effects. Several scales exist to evaluate the potential anticholinergic effect of prescribed drugs scale; but: (i) they are validated in the elderly and mainly assess the cognitive side effect of treat- Psychiatry ments; (ii) they do not concern some of the drugs frequently used in clinical psychiatry in France. The aim of our study is to develop a new scale, the anticholinergic impregnation scale (AIS), with drugs used in France and based on an assessment of the drugs used against peripheral anticholinergic adverse effects. ∗ Corresponding author. Clinical pharmacy service, Établissement public de santé Alsace Nord (EPS Alsace Nord), 141 avenue de Strasbourg, 67170 Brumath, France. -

Fluticasone Furoate, Umeclidinium and Vilanterol (Trelegy)
pat hways Chronic obstructive pulmonary disease: fluticasone furoate, umeclidinium and vilanterol (Trelegy) Evidence summary Published: 14 June 2018 nice.org.uk/guidance/es18 Key points The content of this evidence summary was up-to-date in June 2018. See summaries of product characteristics (SPC), British national formulary (BNF) or the MHRA or NICE websites for up- to-date information. Regulatory status: Fluticasone furoate/umeclidinium/vilanterol (Trelegy, GlaxoSmithKline UK) received a European marketing authorisation in November 2017. This triple-therapy inhaler contains an inhaled corticosteroid (ICS), long-acting beta-2 agonist (LABA) and long-acting muscarinic antagonist (LAMA). It is licensed for maintenance treatment of adults with moderate- to-severe chronic obstructive pulmonary disease (COPD) who are not adequately treated by a combination of an ICS and a LABA (summary of product characteristics). Overview This evidence summary discusses 2 randomised controlled trials (RCTs) looking at the safety and efficacy of fluticasone furoate/umeclidinium/vilanterol (an ICS/LAMA and LABA combination inhaler) in people with COPD who were symptomatic despite regular maintenance treatment and who either had a history, or were at risk, of exacerbations. © NICE 2018. All rights reserved. Subject to Notice of rights (https://www.nice.org.uk/terms-and- Page 1 of conditions#notice-of-rights). 54 Chronic obstructive pulmonary disease: fluticasone furoate, umeclidinium and vilanterol (Trelegy) (ES18) Fluticasone furoate/umeclidinium/vilanterol (Trelegy) is licensed for maintenance treatment of adults with moderate-to severe COPD who are not adequately treated by a combination of an ICS and a LABA. Fluticasone furoate/umeclidinium/vilanterol has been shown to reduce the annual rate of moderate or severe exacerbations by 15% compared with fluticasone furoate/vilanterol.