Genomic Diversification of Giant Enteric Symbionts Reflects Host Dietary Lifestyles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hexosaminidase in Mast Β Primary Role of Degranulation Indicator in Mast Cells?
Does β-Hexosaminidase Function Only as a Degranulation Indicator in Mast Cells? The Primary Role of β-Hexosaminidase in Mast Cell Granules This information is current as of September 25, 2021. Nobuyuki Fukuishi, Shinya Murakami, Akane Ohno, Naoya Yamanaka, Nobuaki Matsui, Kenji Fukutsuji, Sakuo Yamada, Kouji Itoh and Masaaki Akagi J Immunol 2014; 193:1886-1894; Prepublished online 11 July 2014; Downloaded from doi: 10.4049/jimmunol.1302520 http://www.jimmunol.org/content/193/4/1886 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2014/07/11/jimmunol.130252 Material 0.DCSupplemental References This article cites 52 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/193/4/1886.full#ref-list-1 Why The JI? Submit online. by guest on September 25, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2014 by The American Association of -

Quality Assessment Enzyme Analysis for Lysosomal Storage Diseases ERNDIM / EUGT Meeting 5-6 October 2006, Prague
QQuality Assessment Enzyme Analysis for Lysosomal Storage Diseases ERNDIM / EUGT meeting 5-6 October 2006, Prague Results of 1st “large scale” pilot Quality Assessment Enzyme Analysis for Lysosomal Storage Diseases Laboratories Rotterdam Hamburg/Heidelberg Dr, Zoltan Lukacs/Dr. Friederike Bürger QA-pilot for LSD’s the aims • Inter-laboratory variation ─ Many participants: > 30 • Intra-laboratory variation ─ Analyse two pairs of identical control samples (blind) • Proficiency testing of enzyme deficiencies ─ Which samples are best? - Blood samples: most widely used, but impractible - Fibroblasts: good but laborious - EBV lymphoblasts: easy to obtain, not widely used QA-pilot for LSD’s the set up • Samples, without clinical information ─ Leukocytes ─ EBV lymphoblasts ─ Fibroblasts • Enzymes: easy ones ─ 4MU-substrates ─ Simple colorimetric assays • Shipping: economic ─ Send at room temp., postal service - Lyophilised enzymes, stable at room temp. for 5 days - Fibroblasts • Data entry through existing ERNDIM programmes ─ “Metabolite presentation” (www.erndimqa.nl Æ Lysosomal Enzymes) QA-pilot for LSD’s the participants • Questionnaire to members (85 from 21 countries) ─ 40 labs want to join a QA-pilot ─ 65% want to include DBS in future QA-schemes • Samples sent, data returned ─ 40 labs received samples ─ 36 labs entered data on www.erndimqa.nl QA-pilot for LSD’s the samples • 10 samples ─ 4 leukocytes (two duplicate samples) ─ 4 EBV lymphoblasts ─ 2 fibroblasts • 10 easy enzymes ─ specific enzyme activity (e.g.. nmol/h/mg) ─ normalise to -

Sulfite Dehydrogenases in Organotrophic Bacteria : Enzymes
Sulfite dehydrogenases in organotrophic bacteria: enzymes, genes and regulation. Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) an der Universität Konstanz Fachbereich Biologie vorgelegt von Sabine Lehmann Tag der mündlichen Prüfung: 10. April 2013 1. Referent: Prof. Dr. Bernhard Schink 2. Referent: Prof. Dr. Andrew W. B. Johnston So eine Arbeit wird eigentlich nie fertig, man muss sie für fertig erklären, wenn man nach Zeit und Umständen das möglichste getan hat. (Johann Wolfgang von Goethe, Italienische Reise, 1787) DANKSAGUNG An dieser Stelle möchte ich mich herzlich bei folgenden Personen bedanken: . Prof. Dr. Alasdair M. Cook (Universität Konstanz, Deutschland), der mir dieses Thema und seine Laboratorien zur Verfügung stellte, . Prof. Dr. Bernhard Schink (Universität Konstanz, Deutschland), für seine spontane und engagierte Übernahme der Betreuung, . Prof. Dr. Andrew W. B. Johnston (University of East Anglia, UK), für seine herzliche und bereitwillige Aufnahme in seiner Arbeitsgruppe, seiner engagierten Unter- stützung, sowie für die Übernahme des Koreferates, . Prof. Dr. Frithjof C. Küpper (University of Aberdeen, UK), für seine große Hilfsbereitschaft bei der vorliegenden Arbeit und geplanter Manuskripte, als auch für die mentale Unterstützung während der letzten Jahre! Desweiteren möchte ich herzlichst Dr. David Schleheck für die Übernahme des Koreferates der mündlichen Prüfung sowie Prof. Dr. Alexander Bürkle, für die Übernahme des Prüfungsvorsitzes sowie für seine vielen hilfreichen Ratschläge danken! Ein herzliches Dankeschön geht an alle beteiligten Arbeitsgruppen der Universität Konstanz, der UEA und des SAMS, ganz besonders möchte ich dabei folgenden Personen danken: . Dr. David Schleheck und Karin Denger, für die kritische Durchsicht dieser Arbeit, der durch und durch sehr engagierten Hilfsbereitschaft bei Problemen, den zahlreichen wissenschaftlichen Diskussionen und für die aufbauenden Worte, . -

Characterization of Α-L-Fucosidase and Other Digestive Hydrolases From
Acta Tropica 141 (2015) 118–127 Contents lists available at ScienceDirect Acta Tropica journal homepage: www.elsevier.com/locate/actatropica Characterization of ␣-L-fucosidase and other digestive hydrolases from Biomphalaria glabrata Natalia N. Perrella a,b, Rebeca S. Cantinha c,d, Eliana Nakano c, Adriana R. Lopes a,∗ a Laboratory of Biochemistry and Biophysics—Instituto Butantan, São Paulo, Brazil b Programa de Pós Graduac¸ ão Interunidades em Biotecnologia PPIB, Universidade de São Paulo, São Paulo, SP, Brazil c Laboratory of Parasitology—Instituto Butantan, São Paulo, Brazil d Instituto de Pesquisas Energéticas e Nucleares, Universidade de São Paulo, São Paulo, SP, Brazil article info abstract Article history: Schistosoma mansoni is one of the major agents of the disease Schistosomiasis, which is one of the Received 10 February 2014 major global public health concerns. Biomphalaria glabrata is an obligate intermediate mollusc host of Received in revised form 3 July 2014 S. mansoni. Although the development of S. mansoni occurs in the snail hepatopancreas, studies that Accepted 12 August 2014 focus on this organ remain limited. In this study, we biochemically identified five distinct carbohy- Available online 16 September 2014 drases (amylase, maltase, ␣-glucosidase, trehalase, and ␣-L-fucosidase), lipases, and peptidases in the B. glabrata hepatopancreas and focused on the isolation and characterization of the activity of ␣-L- Keywords: fucosidase. The isolated ␣-L-fucosidase has a molecular mass of 141 kDa, an optimum pH of 5.8, and Hepatopancreas ␣ Enzymes is inhibited by Tris, fucose, and 1-deoxyfuconojirimycin. B. glabrata -L-fucosidase is an exoglycosidase ␣-L-Fucosidase that can hydrolyze the natural substrate fucoidan to fucose residues. -

United States Patent (19) 11 Patent Number: 5,981,835 Austin-Phillips Et Al
USOO598.1835A United States Patent (19) 11 Patent Number: 5,981,835 Austin-Phillips et al. (45) Date of Patent: Nov. 9, 1999 54) TRANSGENIC PLANTS AS AN Brown and Atanassov (1985), Role of genetic background in ALTERNATIVE SOURCE OF Somatic embryogenesis in Medicago. Plant Cell Tissue LIGNOCELLULOSC-DEGRADING Organ Culture 4:107-114. ENZYMES Carrer et al. (1993), Kanamycin resistance as a Selectable marker for plastid transformation in tobacco. Mol. Gen. 75 Inventors: Sandra Austin-Phillips; Richard R. Genet. 241:49-56. Burgess, both of Madison; Thomas L. Castillo et al. (1994), Rapid production of fertile transgenic German, Hollandale; Thomas plants of Rye. Bio/Technology 12:1366–1371. Ziegelhoffer, Madison, all of Wis. Comai et al. (1990), Novel and useful properties of a chimeric plant promoter combining CaMV 35S and MAS 73 Assignee: Wisconsin Alumni Research elements. Plant Mol. Biol. 15:373-381. Foundation, Madison, Wis. Coughlan, M.P. (1988), Staining Techniques for the Detec tion of the Individual Components of Cellulolytic Enzyme 21 Appl. No.: 08/883,495 Systems. Methods in Enzymology 160:135-144. de Castro Silva Filho et al. (1996), Mitochondrial and 22 Filed: Jun. 26, 1997 chloroplast targeting Sequences in tandem modify protein import specificity in plant organelles. Plant Mol. Biol. Related U.S. Application Data 30:769-78O. 60 Provisional application No. 60/028,718, Oct. 17, 1996. Divne et al. (1994), The three-dimensional crystal structure 51 Int. Cl. ............................. C12N 15/82; C12N 5/04; of the catalytic core of cellobiohydrolase I from Tricho AO1H 5/00 derma reesei. Science 265:524-528. -

Sporulation Evolution and Specialization in Bacillus
bioRxiv preprint doi: https://doi.org/10.1101/473793; this version posted March 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Research article From root to tips: sporulation evolution and specialization in Bacillus subtilis and the intestinal pathogen Clostridioides difficile Paula Ramos-Silva1*, Mónica Serrano2, Adriano O. Henriques2 1Instituto Gulbenkian de Ciência, Oeiras, Portugal 2Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Oeiras, Portugal *Corresponding author: Present address: Naturalis Biodiversity Center, Marine Biodiversity, Leiden, The Netherlands Phone: 0031 717519283 Email: [email protected] (Paula Ramos-Silva) Running title: Sporulation from root to tips Keywords: sporulation, bacterial genome evolution, horizontal gene transfer, taxon- specific genes, Bacillus subtilis, Clostridioides difficile 1 bioRxiv preprint doi: https://doi.org/10.1101/473793; this version posted March 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Bacteria of the Firmicutes phylum are able to enter a developmental pathway that culminates with the formation of a highly resistant, dormant spore. Spores allow environmental persistence, dissemination and for pathogens, are infection vehicles. In both the model Bacillus subtilis, an aerobic species, and in the intestinal pathogen Clostridioides difficile, an obligate anaerobe, sporulation mobilizes hundreds of genes. -

First Record of Acanthurus Chirurgus (Bloch, 1787) from the Central Mediterranean, with Notes on Other Acanthuridae Recorded in the Region
BioInvasions Records (2017) Volume 6, Issue 2: 105–109 Open Access DOI: https://doi.org/10.3391/bir.2017.6.2.03 © 2017 The Author(s). Journal compilation © 2017 REABIC Rapid Communication A bevy of surgeons: first record of Acanthurus chirurgus (Bloch, 1787) from the central Mediterranean, with notes on other Acanthuridae recorded in the region Julian Evans1,*, Reno Tonna2 and Patrick J. Schembri1 1Department of Biology, University of Malta, Msida MSD2080, Malta 2Namaste Flat 5, Triq il-Merzuq, Birzebbuga, Malta Author e-mails: [email protected] (JE), [email protected] (RT), [email protected] (PJS) *Corresponding author Received: 16 November 2016 / Accepted: 13 December 2016 / Published online: 24 January 2017 Handling editor: Ernesto Azzurro Abstract The doctorfish Acanthurus chirurgus (Bloch, 1787) is reported for the first time from the central Mediterranean, based on a specimen caught in Maltese waters during August 2016. Since the only previous Mediterranean record of this species was based on a single individual observed in the Tyrrhenian Sea, the present record likely represents an independent introduction that may have occurred through the aquarium trade or via shipping. Two other surgeonfish species, Acanthurus coeruleus Bloch and Schneider, 1801 and Acanthurus monroviae Steindachner, 1876, were previously recorded from the central Mediterranean. While A. coeruleus may have established a population in the Levantine Sea, like A. chirurgus it has only been reported once from Malta (and from the central Mediterranean in general); both A. coeruleus and A. chirurgus are, therefore, considered to be casual species in Maltese waters. In contrast, A. monroviae was reported from several Mediterranean countries including Tunisia and Malta in the central Mediterranean. -

First Records of the Fish Abudefduf Sexfasciatus (Lacepède, 1801) and Acanthurus Sohal (Forsskål, 1775) in the Mediterranean Sea
BioInvasions Records (2018) Volume 7, Issue 2: 205–210 Open Access DOI: https://doi.org/10.3391/bir.2018.7.2.14 © 2018 The Author(s). Journal compilation © 2018 REABIC Rapid Communication First records of the fish Abudefduf sexfasciatus (Lacepède, 1801) and Acanthurus sohal (Forsskål, 1775) in the Mediterranean Sea Ioannis Giovos1,*, Giacomo Bernardi2, Georgios Romanidis-Kyriakidis1, Dimitra Marmara1 and Periklis Kleitou1,3 1iSea, Environmental Organization for the Preservation of the Aquatic Ecosystems, Thessaloniki, Greece 2Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, USA 3Marine and Environmental Research (MER) Lab Ltd., Limassol, Cyprus *Corresponding author E-mail: [email protected] Received: 26 October 2017 / Accepted: 16 January 2018 / Published online: 14 March 2018 Handling editor: Ernesto Azzurro Abstract To date, the Mediterranean Sea has been subjected to numerous non-indigenous species’ introductions raising the attention of scientists, managers, and media. Several introduction pathways contribute to these introduction, including Lessepsian migration via the Suez Canal, accounting for approximately 100 fish species, and intentional or non-intentional aquarium releases, accounting for at least 18 species introductions. In the context of the citizen science project of iSea “Is it alien to you?… Share it”, several citizens are engaged and regularly report observations of alien, rare or unknown marine species. The project aims to monitor the establishment and expansion of alien species in Greece. In this study, we present the first records of two popular high-valued aquarium species, the scissortail sergeant, Abudefduf sexfasciatus and the sohal surgeonfish, Acanthurus sohal, in along the Mediterranean coastline of Greece. The aggressive behaviour of the two species when in captivity, and the absence of records from areas close to the Suez Canal suggest that both observations are the result of aquarium intentional releases, rather than a Lessepsian migration. -

Dissimilation of Cysteate Via 3-Sulfolactate Sulfo-Lyase and a Sulfate Exporter in Paracoccus Pantotrophus NKNCYSA
Microbiology (2005), 151, 737–747 DOI 10.1099/mic.0.27548-0 Dissimilation of cysteate via 3-sulfolactate sulfo-lyase and a sulfate exporter in Paracoccus pantotrophus NKNCYSA Ulrike Rein,1 Ronnie Gueta,1 Karin Denger,1 Ju¨rgen Ruff,1 Klaus Hollemeyer2 and Alasdair M. Cook1 Correspondence 1Department of Biology, The University, D-78457 Konstanz, Germany Alasdair Cook 2Institute of Biochemical Engineering, Saarland University, Box 50 11 50, D-66041 [email protected] Saarbru¨cken, Germany Paracoccus pantotrophus NKNCYSA utilizes (R)-cysteate (2-amino-3-sulfopropionate) as a sole source of carbon and energy for growth, with either nitrate or molecular oxygen as terminal electron acceptor, and the specific utilization rate of cysteate is about 2 mkat (kg protein)”1. The initial degradative reaction is catalysed by an (R)-cysteate : 2-oxoglutarate aminotransferase, which yields 3-sulfopyruvate. The latter was reduced to 3-sulfolactate by an NAD-linked sulfolactate dehydrogenase [3?3 mkat (kg protein)”1]. The inducible desulfonation reaction was not detected initially in cell extracts. However, a strongly induced protein with subunits of 8 kDa (a) and 42 kDa (b) was found and purified. The corresponding genes had similarities to those encoding altronate dehydratases, which often require iron for activity. The purified enzyme could then be shown to convert 3-sulfolactate to sulfite and pyruvate and it was termed sulfolactate sulfo-lyase (Suy). A high level of sulfite dehydrogenase was also induced during growth with cysteate, and the organism excreted sulfate. A putative regulator, OrfR, was encoded upstream of suyAB on the reverse strand. Downstream of suyAB was suyZ, which was cotranscribed with suyB. -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

Using Environmental DNA for Marine Monitoring and Planning
Network of Conservation Educators & Practitioners What’s in the Water? Using environmental DNA for Marine Monitoring and Planning Author(s): Kristin E. Douglas, Patrick Shea, Ana Luz Porzecanski, and Eugenia Naro-Maciel Source: Lessons in Conservation, Vol. 10, Issue 1, pp. 29–48 Published by: Network of Conservation Educators and Practitioners, Center for Biodiversity and Conservation, American Museum of Natural History Stable URL: ncep.amnh.org/linc This article is featured in Lessons in Conservation, the official journal of the Network of Conservation Educators and Practitioners (NCEP). NCEP is a collaborative project of the American Museum of Natural History’s Center for Biodiversity and Conservation (CBC) and a number of institutions and individuals around the world. Lessons in Conservation is designed to introduce NCEP teaching and learning resources (or “modules”) to a broad audience. NCEP modules are designed for undergraduate and professional level education. These modules—and many more on a variety of conservation topics—are available for free download at our website, ncep.amnh.org. To learn more about NCEP, visit our website: ncep.amnh.org. All reproduction or distribution must provide full citation of the original work and provide a copyright notice as follows: “Copyright 2020, by the authors of the material and the Center for Biodiversity and Conservation of the American Museum of Natural History. All rights reserved.” Illustrations obtained from the American Museum of Natural History’s library: images.library.amnh.org/digital/ -
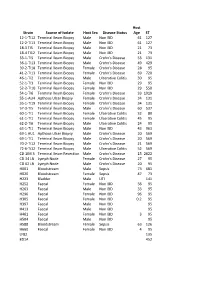
Strain Source of Isolate Host Sex Disease Status Host Age ST
Host Strain Source of Isolate Host Sex Disease Status Age ST 12-1-TI12 Terminal Ileum Biopsy Male Non IBD 61 127 12-2-TI13 Terminal Ileum Biopsy Male Non IBD 61 127 18-3 TI5 Terminal Ileum Biopsy Male Non IBD 21 73 18-4 TI12 Terminal Ileum Biopsy Male Non IBD 21 73 33-1-TI5 Terminal Ileum Biopsy Male Crohn's Disease 53 131 36-1-TI13 Terminal Ileum Biopsy Male Crohn's Disease 49 429 39-2-TI18 Terminal Ileum Biopsy Female Crohn's Disease 28 95 41-2-TI13 Terminal Ileum Biopsy Female Crohn's Disease 69 720 46-1-TI2 Terminal Ileum Biopsy Male Ulcerative Colitis 30 95 52-1-TI3 Terminal Ileum Biopsy Female Non IBD 29 95 52-2-TI10 Terminal Ileum Biopsy Female Non IBD 29 550 54-1-TI6 Terminal Ileum Biopsy Female Crohn's Disease 33 1919 55-1-AU4 Apthous Ulcer Biopsy Female Crohn's Disease 34 131 55-1-TI19 Terminal Ileum Biopsy Female Crohn's Disease 34 131 57-3-TI5 Terminal Ileum Biopsy Male Crohn's Disease 60 537 60-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 32 80 61-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 45 95 62-2-TI6 Terminal Ileum Biopsy Male Ulcerative Colitis 24 95 63-1-TI1 Terminal Ileum Biopsy Male Non IBD 43 963 69-1 AU1 Apthous Ulcer Biopsy Male Crohn's Disease 20 569 69-1-TI1 Terminal Ileum Biopsy Male Crohn's Disease 20 569 70-2-TI12 Terminal Ileum Biopsy Male Crohn's Disease 21 569 72-6-Ti12 Terminal Ileum Biopsy Male Ulcerative Colitis 52 569 CD 1IM 3 Terminal Ileum Resection Male Crohn's Disease 15 2622 CD 34 LN Lymph Node Female Crohn's Disease 27 95 CD 62 LN Lymph Node Male Crohn's Disease 20 95 H001