Nonlinear Effects in Asymmetric Catalysis: a Personal Account Henri B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enantiomers & Diastereomers
Chapter 5 Stereochemistry Chiral Molecules Ch. 5 - 1 1. Chirality & Stereochemistry An object is achiral (not chiral) if the object and its mirror image are identical Ch. 5 - 2 A chiral object is one that cannot be superposed on its mirror image Ch. 5 - 3 1A. The Biological Significance of Chirality Chiral molecules are molecules that cannot be superimposable with their mirror images O O ● One enantiomer NH causes birth defects, N O the other cures morning sickness O Thalidomide Ch. 5 - 4 HO NH HO OMe Tretoquinol OMe OMe ● One enantiomer is a bronchodilator, the other inhibits platelet aggregation Ch. 5 - 5 66% of all drugs in development are chiral, 51% are being studied as a single enantiomer Of the $475 billion in world-wide sales of formulated pharmaceutical products in 2008, $205 billion was attributable to single enantiomer drugs Ch. 5 - 6 2. Isomerisom: Constitutional Isomers & Stereoisomers 2A. Constitutional Isomers Isomers: different compounds that have the same molecular formula ● Constitutional isomers: isomers that have the same molecular formula but different connectivity – their atoms are connected in a different order Ch. 5 - 7 Examples Molecular Constitutional Formula Isomers C4H10 and Butane 2-Methylpropane Cl Cl C3H7Cl and 1-Chloropropane 2-Chloropropane Ch. 5 - 8 Examples Molecular Constitutional Formula Isomers CH O CH C H O OH and 3 3 2 6 Ethanol Methoxymethane O OCH and 3 C H O OH 4 8 2 O Butanoic acid Methyl propanoate Ch. 5 - 9 2B. Stereoisomers Stereoisomers are NOT constitutional isomers Stereoisomers have their atoms connected in the same sequence but they differ in the arrangement of their atoms in space. -

Unit 3 – Stereochemistry
Stereochemistry Stereochemistry refers to the 3-dimensional properties and reactions of molecules. It has its own language and terms that need to be learned in order to fully communicate and understand the concepts. New vocabulary and concepts Handedness Chirality Fischer Projections Depicting Asymmetric Carbons (R) and (S) Nomenclature Enantiomers Diastereomers Optical Activity Stereochemistry Isomers: Different compounds that have the same molecular formula (composition) but different connectivity. Two classes: - Structural (constitutional) isomers: same molecular formula but different bonding sequence - Stereoisomers: same molecular formula, same bonding sequence, but different arrangement in space. Handedness…..Chirality Handedness” right glove doesn’t fit the left hand. Superimposable: A term that describes the ability to precisely overlap one object over another. Only identical objects are superposable, everything else is non-superposable superimposable nonsuperimposable Chiral molecules & Chirality Center Chemical substances can be handed, and they are called chiral. Chiral Molecules: are molecules that are nonsuperimposable on their mirror image. A carbon atom that is bonded to four chiral carbon atom different groups is called chairal carbon atom or stereocenter (asymmetric carbon atom). It is sp3 carbon and labeled with a strict. Achiral: A molecule is achiral if it is superimposable on its mirror image H H Cl Cl Practices on Asymmetric Carbons Example: Identify all asymmetric carbons present in the following compounds. Br Br H OH H H CH2CH3 H C *C C C H Br CH3 * H H H H H H H H H3C Br CH3 * * OH * CH CHCOOH* 3 * * * Fischer Projections: ➢ It is a two-dimensional representation of a three-dimensional organic molecule by projection. -

Chapter 4: Stereochemistry Introduction to Stereochemistry
Chapter 4: Stereochemistry Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane Me Me Me C Me H Bu Bu Me Me 2-methylhexane H H mirror Me rotate Bu Me H 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane H C Et Et Me Pr Pr 3-methylhexane Me Me H H mirror Et rotate H Me Pr 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane .Compounds that are not superimposable with their mirror image are called chiral (in Greek, chiral means "handed") 3-methylhexane is a chiral molecule. .Compounds that are superimposable with their mirror image are called achiral. 2-methylhexane is an achiral molecule. .An atom (usually carbon) with 4 different substituents is called a stereogenic center or stereocenter. Enantiomers Et Et Pr Pr Me CH3 Me H H 3-methylhexane mirror enantiomers Et Et Pr Pr Me Me Me H H Me H H Two compounds that are non-superimposable mirror images (the two "hands") are called enantiomers. Introduction To Stereochemistry Structural (constitutional) Isomers - Compounds of the same molecular formula with different connectivity (structure, constitution) 2-methylpentane 3-methylpentane Conformational Isomers - Compounds of the same structure that differ in rotation around one or more single bonds Me Me H H H Me H H H H Me H Configurational Isomers or Stereoisomers - Compounds of the same structure that differ in one or more aspects of stereochemistry (how groups are oriented in space - enantiomers or diastereomers) We need a a way to describe the stereochemistry! Me H H Me 3-methylhexane 3-methylhexane The CIP System Revisited 1. -

Kinetic Modeling of Chiral Amplification and Enantioselectivity Reversal in Asymmetric Reactions
J. Mex. Chem. Soc. 2007, 51(2), 117-121 Article © 2007, Sociedad Química de México ISSN 1870-249X Kinetic Modeling of Chiral Amplification and Enantioselectivity Reversal in Asymmetric Reactions Jesús Rivera Islas and Thomas Buhse* Centro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, Avenida Universidad 1001, Colonia Chamilpa, 62209 Cuernavaca, Morelos, México. Tel/Fax: +52-7773297997, e-mail: [email protected]. Recibido el 30 de marzo de 2007; aceptado el 18 de mayo de 2007 Abstract: Nonlinear effects in asymmetric synthesis and the occa- Resumen: Los efectos no lineales en síntesis asimétrica y la inver- sionally observed reversal of the enantioselectivity of the employed sión de la enantioselectividad del catalizador empleado son evaluados catalyst are evaluated by a generalized kinetic approach. A non-auto- a través de un modelado cinético generalizado. Un sistema reactivo catalytic and an autocatalytic reaction system are considered as two autocatalítico y otro no autocatalítico son considerados como dos different prototype scenarios. It is predicted that during the course of escenarios prototipos diferentes. Se predice que durante el curso de la enantioselectivity reversal in the non-autocatalytic system a gradual inversión de la enantioselectividad en el sistema no autocatalítico transition between both optically active states under loss of chiral ocurre una transición gradual entre los dos estados óptimamente acti- amplification occurs while in the autocatalytic system a sharp transi- vos donde la amplificación quiral decae mientras en el sistema auto- tion under retention of chiral amplification is observed. catalítico ocurre una transición abrupta observándose la retención de Keywords: Asymmetric synthesis, chiral amplification, nonlinear la amplificación quiral. -

Catalytic Asymmetric Synthesis
Catalytic Asymmetric Synthesis Dr Alan Armstrong Rm 313 (RCS1), ext. 45876; [email protected] 7 lectures Recommended texts: “Catalytic Asymmetric Synthesis, 2nd edition”, ed. I Ojima, Wiley-VCH, 2000 (or 1st ed., 1993, which contains most of the lecture material) “Asymmetric Synthesis”, G Procter, Oxford, 1996 Aims of course: To give students an understanding of the basic principles of asymmetric catalysis, and to demonstrate these in the context of state-of-the-art catalytic asymmetric processes for C-C bond formation and redox processes. Course objectives: At the end of this course you should be able to: • Recognise the types of functional groups which can be prepared by catalytic asymmetric methods discussed in the course; • Use this knowledge in planning the synthesis of enantiomerically enriched compounds from given prochiral starting materials; • Outline the scope and limitations of any methods you propose, with respect to parameters such as turnover, substrate and functional group tolerance, availability of catalysts and/or ligands etc Course content (by lecture, approximately): 1. Introduction and general principles of asymmetric catalysis. Oxidation of functionalised olefins: asymmetric epoxidation of allylic alcohols and enones 2. Oxidation of unfunctionalised olefins: epoxidation, dihydroxylation and aminohydroxylation. 3. Reduction of olefins: asymmetric hydrogenation. 4. Reduction of ketones and imines 5. Asymmetric C-C bond formation: nucleophilic attack on carbonyls, imines and enones 6. Asymmetric transition metal catalysis in C-C bond forming reactions (cross-couplings, Heck reactions, allylic substitution, carbenoid reactions) 7. Chiral Lewis acid catalysis (aldol reactions, cycloadditions, Mannich reactions, allylations) 1 Catalytic Asymmetric Synthesis - Lecture 1 Background and general principles • Why asymmetric synthesis? The need to prepare pharmaceuticals and other fine chemicals as single enantiomers drives the field of asymmetric synthesis. -

Catalytic Enantioselective Desymmetrization of Meso
Catalytic Enantioselective Desymmetrization of Meso Compounds in Total Synthesis of Natural Products: Towards an Economy of Chiral Reagents Jérémy Mérad, Mathieu Candy, Jean-Marc Pons, Cyril Bressy To cite this version: Jérémy Mérad, Mathieu Candy, Jean-Marc Pons, Cyril Bressy. Catalytic Enantioselective Desym- metrization of Meso Compounds in Total Synthesis of Natural Products: Towards an Economy of Chiral Reagents. SYNTHESIS, Georg Thieme Verlag, 2017, 49 (09), pp.1938-1954. 10.1055/s-0036- 1589493. hal-01687264 HAL Id: hal-01687264 https://hal.archives-ouvertes.fr/hal-01687264 Submitted on 18 Jan 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. SYNTHESIS0039-78811437-210X © Georg Thieme Verlag Stuttgart · New York 2017, 49, 1938–1954 1938 short review Syn thesis J. Merad et al. Short Review Catalytic Enantioselective Desymmetrization of Meso Compounds in Total Synthesis of Natural Products: Towards an Economy of Chiral ReaGents OH Jérémy Merad1 HO O Me Mathieu Candy O O Me O Jean-Marc Pons O ( )8 O ( )9 N H H H H Cyril Bressy* MeO Me HO OH -

Absolute Configuration Determination
Chirality in Pharmaceutical Compounds and Absolute Configuration Determination Crystallization: Screening, Application & Process Development X-ray Diffraction Unit Jordi Benet-Buchholz Barcelona, 11 April 2019 Institut Català d’Investigació Química Av. Països Catalans 16 1 43007 Tarragona (Spain) From chiral crystals to the absolute configuration of molecules: Is it an easy way? (–)-Galiellalactone *S OH R * *S O O *R Orthorhombic , P 2 2 2 Absolute configuration: 1 1 1 R1: 3,45 % 4S, 5aR, 7aR, 7bS Flack: 0.01(19) Hooft/Parsons: -0.06(04)/-0.04(05) • F. Nussbaum, R. Hanke, T. Fahrig, J. Benet-Buchholz; Eur. J. Org. Chem. 2004, 2783-2790. 2 Determination of Absolute Configuration of APIs Definitions in Chirality Precedents Methodologies Single Crystal X-ray Structure Determination: Background Candidate samples Sample and Crystal selection Validation and measurements Examples Final schedule 3 Chirality Definitions in Chirality: An object or a system is chiral if it is distinguishable from its mirror image; that is, it cannot be superposed onto it. The word chirality is derived from the Greek word meaning “hand” Paula Benet The most universally recognized example for chirality are the human hands. Live is chiral 4 Chirality Definitions in Chirality: - Each molecule which cannot be superposed with its mirror image is “chiral”. - Molecules with a mirror plane are called “achiral”. The feature that is most often the cause of chirality in molecules is the presence of an asymmetric carbon atom (four different substituents) Two mirror images of a chiral molecule are called enantiomers or optical isomers Foto cristall papallona 5 Conglomerate of crystals forming enantiomeric butterflies Chirality Definitions in Chirality: Pairs of enantiomers are often designated as “right- or left-handed”. -

Chirality: the Handedness of Molecules
06 Chirality: The Handedness of Molecules Tartaric acid is found in grapes and other fruits, both free and as its salts (see Section 6.4B). Inset: A model of tartaric acid. (© fatihhoca/iStockphoto) KEY QUESTIONS 6.1 What Are Stereoisomers? 6.9 What Is the Significance of Chirality in the 6.2 What Are Enantiomers? Biological World? 6.3 How Do We Designate the Configuration of a 6.10 How Can Enantiomers Be Resolved? Stereocenter? HOW TO 6.4 What Is the 2n Rule? 6.1 How to Draw Enantiomers 6.5 How Do We Describe the Chirality of 6.2 How to Determine the R & S Configuration Cyclic Molecules with Two Stereocenters? without Rotating the Molecule 6.6 How Do We Describe the Chirality 6.3 How to Determine Whether Two Compounds Are of Molecules with Three or More the Same, Enantiomers, or Diastereomers without Stereocenters? the Need to Spatially Manipulate the Molecule 6.7 What Are the Properties of Stereoisomers? 6.8 How Is Chirality Detected in the CHEMICAL CONNECTIONS Laboratory? 6A Chiral Drugs IN THIS CHAPTER, we will explore the relationships between three-dimensional ob- jects and their mirror images. When you look in a mirror, you see a reflection, or mirror Mirror image The reflection image, of yourself. Now, suppose your mirror image becomes a three-dimensional object. of an object in a mirror. 167 168 CHAPTER 6 Chirality: The Handedness of Molecules We could then ask, “What is the relationship between you and your mirror image?” By relationship, we mean “Can your reflection be superposed on the original ‘you’ in such a way that every detail of the reflection corresponds exactly to the original?” The answer is that you and your mirror image are not superposable. -

Stereochemistry
Stereochemistry For Cheminformatics Introduction The branch of chemistry treating of the space relations of atoms in molecules. Definition • The branch of chemistry that deals with spatial arrangements of atoms in molecules and the effects of these arrangements on the chemical and physical properties of substances. • The study of the three-dimensional arrangement of atoms or groups within molecules and the properties which follow from such arrangement. The importance of stereochemistry • The importance of stereochemistry in drug action is gaining greater attention in medical practice, and a basic knowledge of the subject will be necessary for clinicians to make informed decisions regarding the use of single-enantiomer drugs. Many of the drugs currently used in psychiatric practice are mixtures of enantiomers. For some therapeutics, single-enantiomer formulations can provide greater selectivities for their biological targets, improved therapeutic indices, and/or better pharmacokinetics than a mixture of enantiomers. Importance of stereochemistry • Many biologically active synthetic drugs contain chiral centers, although they are used as racemic mixtures. Enantiomers are hard to distinguish in the chemical laboratory but are readily discriminated in the body and differ in their biological activities and disposition. The pharmacokinetic profiles of enantiomers can be variable, especially for drugs with a first-pass effect and enantioselective pharmacokinetic monitoring should be carried out. Ultimately, whether to exploit a racemate or a single enantiomer in therapy is a multi-faceted decision to which drug disposition data have important contributions to make. Stereoisomers and stereoisomerism • Molecules that have identical molecular structures but differ in the relative spatial arrangement of component parts are stereoisomers. -
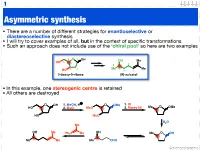
Asymmetric Synthesis
1 Asymmetric synthesis • There are a number of different strategies for enantioselective or diastereoselective synthesis • I will try to cover examples of all, but in the context of specific transformations • Such an approach does not include use of the ‘chiral pool’ so here are two examples 4 O 1 OH OH Me HO 5 5 2 3 2 Me 4 Me HO 3 1 2-deoxy-D-ribose (R)-sulcatol • In this example, one stereogenic centre is retained • All others are destroyed O OH 1. MeOH, H O OMe 1. KI HO 2. MsCl MsO 2. Raney Ni Me O OMe HO MsO H2O Me OH Me OH Ph3P Me Me O OH Me Me Me CHO Advanced organic 2 ‘Chiral pool’ II Me Me remove OH Me Me stereogenic 3 steps O O centre HO 2 4 OH PCC 3 O OH O O 6 OH HO 1 O 5 OTBDPS OTBDPS D-mannose BnO O BnO O addition of 1. NaBH protecting groups overall retention of 4 stereochemistry 2. Tf2O stereoselective Me 1. TBAF Me Me reduction Me 2. PCC Me Me O O O 3. Ph P=CHCHO NaN O 3 O 3 O N3 N3 OTf OTBDPS OTBDPS BnO O CHO BnO O BnO O Pd / C hydrogenolysis of benzyl (Bn) group & H reductive amination of resultant 2 aldehyde Me HO two step reversal of Me O 2 1 stereogenic centre reduction 1. H2, Pd / C, H of alkene & H H 3 O N 2. TFAA azide HO 4 N followed by reductive H 6 amination BnO O 5 H HO swainsonine • In this example three stereogenic centres are retained • One stereogenic centre undergoes multiple inversion -- but overall it is retained Advanced organic 3 Stereoselective reactions of alkenes • Alkenes are versatile functional groups that, as we shall see, present plenty of scope for the introduction of stereochemistry • Hydroboration permits the selective introduction of boron (surprise), which itself can undergo a wide-range of stereospecific reactions Substrate control Me H H Me H H Me 1. -

A Scan of My Lecture Notes from Today's Lecture
We will Oost an announcement and send an email with exam instructions specific book Open note open Zoom a TA proctoring by have two hours You will on 7 9 pm Thursday have 20 additional You will minutes to upload using Gradescope We have a Zoom will hotline number if phone have you technology issues or accommodations Specif alternative exam information are is If you coming extra time you receiving will at 7 pm and start later go THE UT HONOR APPLIES CODE The rule B cannot only you collaborate with each other There is a review session tomorrow frm evening 5 7 PM STEREOCHEMISTRY HOPEFULLY MADE SIMPLER Stereoisomers-molecules that have the same constitution, but different disposition of groups in space. In other words the atoms are connected to each other in the same way, they only differ with respect to relative orientation in three--dimensional space. Chiral-General Definition-Any object that is not superimposable on its mirror image. Your hands are chiral, that is why you need two different leather gloves, one that only fits your right hand, and one that only fits your left hand. If your hands were superimposable, then you would only need one kind of glove and it would fit both hands. Chiral-Chemistry Definition; Atom-Any tetrahedral carbon atom that has four different substituents is a chiral center. Any tetrahedral carbon atom that has four different substituents is a chiral center (it was worth repeating). This is a simple consequence of geometry; there are two different ways to place four different substituents in a tetrahedral arrangement. -

Stereochemistry
Lecture note- 2 Organic Chemistry CHE 502 STEREOCHEMISTRY DEPARTMENT OF CHEMISTRY UTTARAKHAND OPEN UNIVERSITY UNIT 4: STEREOCHEMISTRY 1 CONTENTS 4.1 Objectives 4.2 Introduction 4.3 Isomerism 4.4 Structural (Constitutional) Isomerism 4.5 Stereo (Configurational) isomerism 4.5.1 Geometrical Isomerism 4.5.2 Optical Isomerism 4.6 Element of Symmetry 4.7 Stereogenic centre (Stereogenicity) 4.7.1 Optical activity and Enantiomerism 4.7.2 Properties of enantiomerism 4.7.3 Chiral and achiral molecules with two stereogenic centers 4.7.4 Diastereomers 4.7.5 Properties of Diastereomers 4.7.6 Erythro (syn) Threo (anti) diastereomers 4.7.7 Meso compounds 4.8 Relative and absolute configurations 4.8.1 D/L nomenclature 4.8.2 R/S nomenclature 4.8.3 Sequence Rule 4.9 Newman and sawhorse projection formulae 4.10 Fisher flying and wedge formulae 4.11 Racemic mixture (racemates) 4.12 Quasi enantiomers 4.13 Quasi racemates 4.14 Stereochemistry of allenes, spiranes, biphenyls, ansa compounds, cyclophanes and related compounds 4.15 Summary 4.16 Terminal Questions 4.17 Answers 4.1 OBJECTIVES In this unit learner will be able to: ➢ Depict various types of isomerism exhibited by organic compounds and their representation ➢ Analyze the three dimensional depictions of organic compounds and their two dimensional representations. ➢ Learn Stereogenicity, chirality, enantiomerism, diastereomerism, their relative and absolute configurations ➢ Learn about the various stereo chemical descriptors such as (cis-trans, E/Z, D/L, d/l, erythro/threo, R/S and syn/anti) given to organic molecules differ ➢ Describe the stereochemistry of various rigid and complex molecules like spiranes, adamentanes, catenanes, cyclophanes etc.