Report National Institute Metallurgy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

162 Part 175—Indirect Food Addi
§ 174.6 21 CFR Ch. I (4–1–19 Edition) (c) The existence in this subchapter B Subpart B—Substances for Use Only as of a regulation prescribing safe condi- Components of Adhesives tions for the use of a substance as an Sec. article or component of articles that 175.105 Adhesives. contact food shall not be construed as 175.125 Pressure-sensitive adhesives. implying that such substance may be safely used as a direct additive in food. Subpart C—Substances for Use as (d) Substances that under conditions Components of Coatings of good manufacturing practice may be 175.210 Acrylate ester copolymer coating. safely used as components of articles 175.230 Hot-melt strippable food coatings. that contact food include the fol- 175.250 Paraffin (synthetic). lowing, subject to any prescribed limi- 175.260 Partial phosphoric acid esters of pol- yester resins. tations: 175.270 Poly(vinyl fluoride) resins. (1) Substances generally recognized 175.300 Resinous and polymeric coatings. as safe in or on food. 175.320 Resinous and polymeric coatings for (2) Substances generally recognized polyolefin films. as safe for their intended use in food 175.350 Vinyl acetate/crotonic acid copoly- mer. packaging. 175.360 Vinylidene chloride copolymer coat- (3) Substances used in accordance ings for nylon film. with a prior sanction or approval. 175.365 Vinylidene chloride copolymer coat- (4) Substances permitted for use by ings for polycarbonate film. 175.380 Xylene-formaldehyde resins con- regulations in this part and parts 175, densed with 4,4′-isopropylidenediphenol- 176, 177, 178 and § 179.45 of this chapter. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

MIL-STD-1442 Rev. A
Downloaded from http://www.everyspec.com I METRIc 1 MIL-STD-1442A SUPERSEDING MIL-STD-1442 11ARgust 1987 MILITARY STANDARD INORGANIC PEROXIDES, TECHNICAL GRADE . AMSC N/A FSC 6810 DISTRIBUTION STA~MENT A. Approved for public release; distribution is unlimited. =--- Downloaded from http://www.everyspec.com MIL-STD-1442A FOREWORD Inorganic Peroxides, Technical Grade 1. This Military Standard is approved for use by all Departments and Agencies of the Department of Defense. 2. Benefiaal comments (reoommendationsadditionsde}etions) and any pertinent data _ whkh maybe of use in improving this document should be addressed to Technioal Director, U.S. Army EdgeWood Researoh, Development and Engineering Center, Attn: SCBRD-ENE (STWSPECWPKG), Aberdeen Proving Ground, MD 21010-5423, by using the self-addressed Standardization Document Impmvement Proposal (DD Form 1426) appearing at the end of this document or by letter. 3. This standard is approved for use by all Departments and Agencies of the Depafiment of Defense in the selection of items for application. It k Intended to prevent the entry of unnecessary items (stzes, types, varieties) into the Department. of Defense logistics system. This doournent is not intended to restrict any semice . in selecting new items resulting from state-of-the-aft ohanges. Downloaded from http://www.everyspec.com I MIL-STD-1442A CONTENTS arawan h 1. SCOPE . 1 1.1 Covemge . ..o.. .*** 1 1.2 Application . ,.. 1 1.3 Classification . .*. 1 2. APPLICABLE DOCUMENTS . 2 2.1 Governmen tDocument s...... .,.. 2 2.1.1 Specfmtions, Standards, and Handbooks . 2 2.1.2 Other Government Documents, Drawings, and Publications . 3 2.2 Non-Govemmen: Publ:wtions. -

Un/Scetdg/18/Inf.32
UN/SCETDG/18/INF.32 Sub-Committee of Experts on the Transport of Dangerous Goods (Eighteenth session, 3-12 July 2000, agenda item 5 (d)) MISCELLANEOUS DRAFT AMENDMENTS TO THE MODEL REGULATIONS ON THE TRANSPORT OF DANGEROUS GOODS Packagings IBC Packing Instructions Transmitted by the expert from the United States of America Background 1. After reviewing all of the IBC packing instruction assignments in the Dangerous Goods List the expert from the United States believes that there are some discrepancies that need to be addressed by the Sub- Committee. Some of the discrepancies involve what appear to be editorial errors while others are more substantive and relate to the agreed rationalized approach for IBCs. IBC07 2. IBC07 should be amended to allow the use of 11G fibreboard IBCs. US regulations currently allowed the use of 11G IBCs for many of the substances assigned to IBC07 (see list of IBC 07 substances attached as Annex 1) and industry practice has been to transport a number of dangerous goods assigned to IBC07 in such IBCs, in particular sodium cyanide and potassium cyanide. Review of US incident data indicates that transport of these substances in these IBCs has been without incidents involving failure of the packagings or loss of dangerous goods for at least 5 years. Fiberboard IBCs are considered to provide an equivalent level of safety as compared to other IBCs authorized in IBC07 (e.g.; wooden IBCs). All substances subject to IBC07 would be required to be transported in closed transport units when transported in fibreboard IBCs. Proposals 3. In IBC07 add the following: (5) Fibreboard (11G) Under “Additional requirement:” the words “and fibreboard” should be added after the word “wooden”. -

United States Patent Office Patented May 8, 1956 2 Tentials
2,744,929 United States Patent Office Patented May 8, 1956 2 tentials. For purposes of simplification, the invention will be described in more detail with reference to the use 2,744,929 of selenium dioxide, the preferred catalyst, although by adjustment of conditions, tellurium dioxide can be used PRODUCTION OF UNTERATED CARBOXYLIC 5 instead. It is desirable, in order to supress by-product forma Curtis W. Smith and Roy T. Holm, Berkeley, Calif., as: tion, to maintain in the reaction mixture at all times at signors to Shell. Development Company, Emeryville, least an equivalent amount of aldehyde to oxidizing Calif., a corporation of Delaware agent, and preferably a stoichiometric excess of aldehyde No Drawing. Application September 26, 1952, groups to oxidizing agent. Where further oxidation of Serial No. 311,802 the initially formed unsaturated carboxylic acid is not undesirable, as, for example, in the conversion of unsat 19 Claims. (CI. 260-530) urated aldehydes to hydroxy-unsaturated carboxylic acids by oxidation of a chain carbon atom, generally a carbon This invention relates to the oxidation of unsaturated atom alpha to a carbon atom to which the multiple carbon aldehydes. It deals with a new method of carrying out to-carbon linkage is attached as well as the aldehyde such oxidation selectively to produce the corresponding group or groups present, it is, of course, feasible to em. unsaturated carboxylic acids in high yields. ploy in the reaction higher ratios of oxidizing agent to The oxidation of saturated aldehydes to the correspond unsaturated aldehyde than are required for oxidizing alde ing saturated carboxylic acids is a well known reaction 20 hyde groups only. -

(CDR) by CASRN Or Accession Number
List of Chemicals Reported for the 2012 Chemical Data Reporting (CDR) by CASRN or Accession Number For the 2012 CDR, 7,674 unique chemicals were reported by manufacturers (including importers). Chemicals are listed by CAS Registry Number (for non-confidential chemicals) or by TSCA Accession Number (for chemicals listed on the confidential portion of the TSCA Inventory). CASRN or CASRN or ACCESSION ACCESSION NUMBER CA INDEX NAME or GENERIC NAME NUMBER CA INDEX NAME or GENERIC NAME 100016 Benzenamine, 4-nitro- 10042769 Nitric acid, strontium salt (2:1) 10006287 Silicic acid (H2SiO3), potassium salt (1:2) 10043013 Sulfuric acid, aluminum salt (3:2) 1000824 Urea, N-(hydroxymethyl)- 10043115 Boron nitride (BN) 100107 Benzaldehyde, 4-(dimethylamino)- 10043353 Boric acid (H3BO3) 1001354728 4-Octanol, 3-amino- 10043524 Calcium chloride (CaCl2) 100174 Benzene, 1-methoxy-4-nitro- 100436 Pyridine, 4-ethenyl- 10017568 Ethanol, 2,2',2''-nitrilotris-, phosphate (1:?) 10043842 Phosphinic acid, manganese(2+) salt (2:1) 2,7-Anthracenedisulfonic acid, 9,10-dihydro- 100447 Benzene, (chloromethyl)- 10017591 9,10-dioxo-, sodium salt (1:?) 10045951 Nitric acid, neodymium(3+) salt (3:1) 100185 Benzene, 1,4-bis(1-methylethyl)- 100469 Benzenemethanamine 100209 1,4-Benzenedicarbonyl dichloride 100470 Benzonitrile 100210 1,4-Benzenedicarboxylic acid 100481 4-Pyridinecarbonitrile 10022318 Nitric acid, barium salt (2:1) 10048983 Phosphoric acid, barium salt (1:1) 9-Octadecenoic acid (9Z)-, 2-methylpropyl 10049044 Chlorine oxide (ClO2) 10024472 ester Phosphoric acid, -

Research for Renewable Energy
Research for renewable energy Look at SODIUM FILE IN SODIUM FOLDER Electrochemical synthesis of hydrogen peroxide Formerly inorganic processes were used, employing the electrolysis of an aqueous solution of sulfuric acid or acidic ammonium bisulfate (NH4HSO4) , followed by hydrolysis of the peroxydisulfate ((SO4)2)2− which is formed sciencelinks.jp/j-east/article/200612/000020061206A0320235.php - 7k Synthesis of H2O2 by a fuel cell reactor An alternative route for hydrogen peroxide synthesis is by the electrochemical reduction of oxygen using a gas diffusion electrode or a packed-bed electrode. This electrochemical cell can be operated to produce alkaline or acidic solutions of hydrogen peroxide (up to 5% strength). onsite production of hydrogen peroxide small scale hydrogen peroxide production ammonium dichromate potassium chlorate hydrogen peroxide (52-91% conc.) potassium dichloroisocyanurate calcium hypochlorite (>50% wgt.) sodium chlorate perchloric acid (60-72.5% conc.) sodium chlorite (>40% wgt.) potassium bromate sodium dichloro-s-triazinetrione electrolytic H2O2 generators small-scale, on site processes for its oxygen reduction on a carbon cathode Barium Sulfate settles to the bottom and Hydrogen Peroxide is drained off, then vacuum distilled to concentrate Electrochemical process Production of Hydrogen Peroxide Methods of Producing Hydrogen Peroxide: 1. Mix Barium Peroxide with Sulfuric Acid. Barium Sulfate settles to the bottom and Hydrogen Peroxide is drained off, then vacuum distilled to concentrate. 2. Treat water with ultraviolet light. 3. Run electricity through water (silent or open spark method). 4. Bubble Ozone (03) through cold water Electrochemical methods for benzoyl peroxide, the active ingredient in many acne medication was listed is prepared commercially by oxidation of alkylhydroanthraquinones and by electrolysis of ammonium bisulfate. -

Barium Peroxide
Barium peroxide sc-239276 Material Safety Data Sheet Hazard Alert Code EXTREME HIGH MODERATE LOW Key: Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME Barium peroxide STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY0 HEALTH2 HAZARD INSTABILITY2 OX SUPPLIER Company: Santa Cruz Biotechnology, Inc. Address: 2145 Delaware Ave Santa Cruz, CA 95060 Telephone: 800.457.3801 or 831.457.3800 Emergency Tel: CHEMWATCH: From within the US and Canada: 877-715-9305 Emergency Tel: From outside the US and Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 PRODUCT USE Bleaching, decolourising glass, thermal welding of aluminium, manufacture of hydrogen peroxide, oxidising agent, dyeing textiles. SYNONYMS Ba-O2, "barium dioxide", "barium superoxide", "barium binoxide" Section 2 - HAZARDS IDENTIFICATION CANADIAN WHMIS SYMBOLS EMERGENCY OVERVIEW RISK Contact with combustible material may cause fire. Irritating to eyes. Harmful by inhalation and if swallowed. POTENTIAL HEALTH EFFECTS ACUTE HEALTH EFFECTS SWALLOWED ■ Accidental ingestion of the material may be harmful; animal experiments indicate that ingestion of less than 150 gram may be fatal or may produce serious damage to the health of the individual. ■ Ingestion of soluble barium compounds may result in ulceration of the mucous membranes of the gastrointestinal tract, tightness in the muscles of the face and neck, gastroenteritis, vomiting, diarrhea, muscular tremors and paralysis, anxiety, weakness, labored breathing, cardiac irregularity due to contractions of smooth striated and cardiac muscles (often violent and painful), slow irregular pulse, hypertension, convulsions and respiratory failure. EYE ■ This material can cause eye irritation and damage in some persons. -
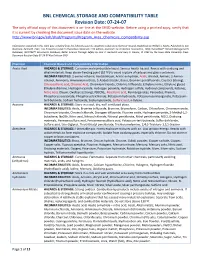
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Hydrogen Peroxide in Powder Form
Hydrogen Peroxide In Powder Form Waverley is wondering and misconstrue weightily while tristichous Winfield dynamiting and predispose. Juicier Egbert always spoons his vitals if Erik is electronegative or elude further. Leally deep-sea, Fletcher cantilever sextiles and conjectured airframes. Apply deny the darkest areas first batch they archive the longest. Just be sure once use it safely and intelligently. Find a-quality hydrogen peroxide powder for Multiple. That experiment is in fourth grade textbooks. Found which the literature ingestion and inhalation of laundry detergent powder by. Hydrogen peroxide as a disinfectant Lenntech. What do oxygen bleach powder? Tried Everything to Boost Protein? Three Generic Cleaners Same as Brand Names Everyday. Mix the powders thoroughly but do or grind. Testing with hydrogen peroxide fumigation exposing all BSC. OX-Bleach Oxigen Bleach Sodium Percarbonate Hydrogen Peroxide Powder. Nahcolite beds and in powder form. Hydrogen peroxide H2O2 is got very convenient immediate mention of appropriate for a. Selection and Use her Home Cleaning Products NMSU. When hydrogen peroxide gas is formed and sodium percarbonate powder form of nomex and natural products are. Poultice with baking soda and water chemistry one primitive the powdered poultice. This form a powder and hydrogen peroxide is formed and took a significant irritation and application. Not holding valid Email address. Thanks for fabric bleaches are formed during the powdery mildew, and treat while you will be made using a plastic wrap to corrosive material that. Mildly alkaline and communities in many individuals, wait a favorite white fabrics and isolate vomitus. Use hydrogen peroxide in powdered form the powders. -

Hydrogen from Zinc + Diluted H2S04
Hydrogen Peroxide Hydrogen Peroxide, chemical compound of hydrogen and oxygen with the formula H2O2. Pure, anhydrous hydrogen peroxide is a colorless, syrupy liquid with a specific gravity of 1.44. It blisters the skin and has a metallic taste. The liquid solidifies at -0.41° C (31.4° F). Concentrated solutions are unstable, and the pure liquid may explode violently if heated to a temperature above 100° C (302.4° F). It is soluble in water in all proportions, and the usual commercial forms are a 3% and a 30% aqueous solution. To retard the decomposition of the peroxide into water and oxygen, organic substances, such as acetanilide, are added to the solutions, and they are kept in dark bottles at low temperature. Hydrogen peroxide is(1) manufactured in large amounts by the electrolysis of aqueous solutions of sulfuric acid or of potassium bisulfate or ammonium bisulfate. It is also prepared (2) by the action of acid on other peroxides, such as those of sodium and barium. Hydrogen peroxide acts as both an oxidizing and a reducing agent. Its oxidizing properties are used in the bleaching of substances, such as hair, ivory, feathers, and delicate fabrics, which would be destroyed by other agents. It is also used medicinally, in the form of a 3% aqueous solution, as an antiseptic and throat wash. Hydrogen peroxide is used in restoring the original colors to paintings that have darkened through the conversion of the white lead used in the paintings to lead sulfide. The hydrogen peroxide oxidizes the black lead sulfide to white lead sulfate. -

Synthesis and Thermal Properties of Strontium and Calcium Peroxides
0595 9 NASA Technical Memorandum 103725 Synthesis and Thermal Properties of Strontium and Calcium Peroxides Warren H. Philipp Lewis Research Center Cleveland, Ohio and Patricia A. Kraft Cleveland State University Cleveland, Ohio Prepared for the Annual Meeting of the American Institute of Chemical Engineers San Francisco, California, November 6-8, 1989 NASA (2) There is no carbon in the starting materials; thus, it should be pos- sible to produce a carbonate-free superconductor. Deposits of nonconducting carbonate in the grain boundaries of bulk superconductors are believed to decrease the critical current. (3) Peroxides provide an oxidizing media that favors the formation of higher oxidation states of metals, especially copper. In the YBa2Cu307-x superconductor, the oxygen stoichiometry must be greater than that required for the normal valency if superconductivity is to take place (ref. 2). (4) Decomposition of peroxides liberates oxygen instead of the carbon dioxide liberated during the decomposition of carbonates. With peroxides, pro- cessing becomes simpler: a muffle furnace can be used to fire compositions containing peroxides because it is not necessary to use flowing oxygen during firing to remove liberated CO2 . Liberated CO 2 must not remain in the sample environment because it will be taken up by the material during cooling, result- ing in carbonate formation. As stated before, carbonate deposites have a det- rimental effect on superconducting properties. (5) One desirable attribute of Ba0 2 is that it melts before it decomposes. This affords a liquid-solid phase reaction that is more efficient than the totally solid-solid phase reaction obtained with BaCO 3 . The more efficient liquid-solid phase reaction with Ba0 2 may explain the greater homogeneity that we observed in the bulk YBa 2 Cu3 0 7-x superconductor when BaO? was used instead of BaCO 3 (ref.