A CREB3–ARF4 Signalling Pathway Mediates the Response to Golgi Stress and Susceptibility to Pathogens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Driver Strength Index Highlights Important Cancer Genes in TCGA Pancanatlas Patients
medRxiv preprint doi: https://doi.org/10.1101/2021.08.01.21261447; this version posted August 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Novel Driver Strength Index highlights important cancer genes in TCGA PanCanAtlas patients Aleksey V. Belikov*, Danila V. Otnyukov, Alexey D. Vyatkin and Sergey V. Leonov Laboratory of Innovative Medicine, School of Biological and Medical Physics, Moscow Institute of Physics and Technology, 141701 Dolgoprudny, Moscow Region, Russia *Corresponding author: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2021.08.01.21261447; this version posted August 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Abstract Elucidating crucial driver genes is paramount for understanding the cancer origins and mechanisms of progression, as well as selecting targets for molecular therapy. Cancer genes are usually ranked by the frequency of mutation, which, however, does not necessarily reflect their driver strength. Here we hypothesize that driver strength is higher for genes that are preferentially mutated in patients with few driver mutations overall, because these few mutations should be strong enough to initiate cancer. -

A Potent and Selective Inhibitor of Cdc42 Gtpase Zurab Surviladze University of New Mexico
Cedarville University DigitalCommons@Cedarville Pharmacy Faculty Publications School of Pharmacy 2010 A Potent and Selective Inhibitor of Cdc42 GTPase Zurab Surviladze University of New Mexico Anna Waller University of New Mexico J. Jacob Strouse University of New Mexico Cristian G. Bologa University of New Mexico Oleg Ursu University of New Mexico See next page for additional authors Follow this and additional works at: http://digitalcommons.cedarville.edu/pharmacy_publications Part of the Medicinal and Pharmaceutical Chemistry Commons Recommended Citation 2010: Surviladze, Z., Waller A., Strouse, J., Bologa, C., & Ursu, O., A Potent and Selective Inhibitor of Cdc42 GTPase, submitted to National Institutes of Health. This Web Publication is brought to you for free and open access by DigitalCommons@Cedarville, a service of the Centennial Library. It has been accepted for inclusion in Pharmacy Faculty Publications by an authorized administrator of DigitalCommons@Cedarville. For more information, please contact [email protected]. Authors Zurab Surviladze, Anna Waller, J. Jacob Strouse, Cristian G. Bologa, Oleg Ursu, Virginia M. Salas, Genevieve K. Phillips, John F. Parkinson, Elsa Romero, Angela Wandinger-Ness, Larry A. Sklar, Chad E. Schroeder, Denise S. Simpson, Julica Nöth, Jenna Wang, Jennifer E. Golden, and Jeffrey Aubé This web publication is available at DigitalCommons@Cedarville: http://digitalcommons.cedarville.edu/pharmacy_publications/19 Probe Report Title: A Potent and Selective Inhibitor of Cdc42 GTPase Authors: UNM Center for Molecular Discovery: Zurab Surviladze, Anna Waller, J. Jacob Strouse, Cristian Bologa, Oleg Ursu, Virginia Salas, John F. Parkinson, Genevieve K. Phillips, Elsa Romero, Angela Wandinger-Ness and Larry A. Sklar. Kansas University Specialized Chemistry Center: Chad Schroeder, Denise Simpson, Julica Nöth, Jenna Wang, Jennifer Golden, and Jeffrey Aubé. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer
cancers Review An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer Claire M. Robinson 1,† , Aaron Talty 1,†, Susan E. Logue 2,3 , Katarzyna Mnich 1, Adrienne M. Gorman 1 and Afshin Samali 1,* 1 Apoptosis Research Centre, School of Natural Sciences, National University of Ireland, H91 W2TY Galway, Ireland; [email protected] (C.M.R.); [email protected] (A.T.); [email protected] (K.M.); [email protected] (A.M.G.) 2 Department of Human Anatomy and Cell Science, Rady Faculty of Health Sciences, Max Rady College of Medicine, University of Manitoba, Winnipeg, MB R3E 0J9, Canada; [email protected] 3 Research Institute in Oncology and Hematology, Cancer Care Manitoba, Winnipeg, MB R3E 0V9, Canada * Correspondence: [email protected] † These authors contributed equally to this work. Simple Summary: Pancreatic cancer refers to a group of malignancies of which pancreatic ductal adenocarcinoma (PDAC) is the most common form. It is an aggressive tumour with few treatment options and very poor outcomes. There is an unmet need for novel targeted therapies for PDAC. In this regard, a better understanding of PDAC biology and in particular new pathways that contribute to disease progression would help identify novel targets for therapeutic intervention. Growing evidence implicates the unfolded protein response (UPR) in many cancers, and this article explores the evidence that supports a role for the UPR in PDAC. Abstract: Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer and one of the leading causes of cancer-associated deaths in the world. It is characterised by dismal response rates to conventional therapies. -

Comprehensive Biological Information Analysis of PTEN Gene in Pan-Cancer
Comprehensive biological information analysis of PTEN gene in pan-cancer Hang Zhang Shanghai Medical University: Fudan University https://orcid.org/0000-0002-5853-7754 Wenhan Zhou Shanghai Medical University: Fudan University Xiaoyi Yang Shanghai Jiao Tong University School of Medicine Shuzhan Wen Shanghai Medical University: Fudan University Baicheng Zhao Shanghai Medical University: Fudan University Jiale Feng Shanghai Medical University: Fudan University Shuying Chen ( [email protected] ) https://orcid.org/0000-0002-9215-9777 Primary research Keywords: PTEN, correlated genes, TCGA, GEPIA, UALCAN, GTEx, expression, cancer Posted Date: April 12th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-388887/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/21 Abstract Background PTEN is a multifunctional tumor suppressor gene mutating at high frequency in a variety of cancers. However, its expression in pan-cancer, correlated genes, survival prognosis, and regulatory pathways are not completely described. Here, we aimed to conduct a comprehensive analysis from the above perspectives in order to provide reference for clinical application. Methods we studied the expression levels in cancers by using data from TCGA and GTEx database. Obtain expression box plot from UALCAN database. Perform mutation analysis on the cBioportal website. Obtain correlation genes on the GEPIA website. Construct protein network and perform KEGG and GO enrichment analysis on the STRING database. Perform prognostic analysis on the Kaplan-Meier Plotter website. We also performed transcription factor prediction on the PROMO database and performed RNA-RNA association and RNA-protein interaction on the RNAup Web server and RPISEq. -
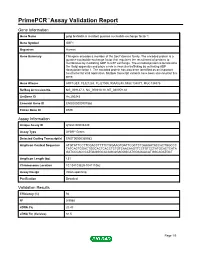
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name golgi brefeldin A resistant guanine nucleotide exchange factor 1 Gene Symbol GBF1 Organism Human Gene Summary This gene encodes a member of the Sec7 domain family. The encoded protein is a guanine nucleotide exchange factor that regulates the recruitment of proteins to membranes by mediating GDP to GTP exchange. The encoded protein is localized to the Golgi apparatus and plays a role in vesicular trafficking by activating ADP ribosylation factor 1. The encoded protein has also been identified as an important host factor for viral replication. Multiple transcript variants have been observed for this gene. Gene Aliases ARF1GEF, FLJ21263, FLJ21500, KIAA0248, MGC134877, MGC134878 RefSeq Accession No. NG_008147.1, NC_000010.10, NT_030059.13 UniGene ID Hs.290243 Ensembl Gene ID ENSG00000107862 Entrez Gene ID 8729 Assay Information Unique Assay ID qHsaCID0008440 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369983 Amplicon Context Sequence ATGTATTCCTTCGACCTTTTCTGGAAGTGATTCGCTCTGAAGATACCACTGGCCC TATCACTGGACTGGCACTCACCTCTGTCAACAAGTTCCTGTCCTATGCACTCATA GATCCCACCCATGAGGGCACAGCAGAGGGCATGGAGAACATGGCAGATGCT Amplicon Length (bp) 131 Chromosome Location 10:104103829-104111062 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 94 R2 0.9988 cDNA Cq 20.43 cDNA Tm (Celsius) 84.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 36.2 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay -

Brefeldin A, a Cytotoxin Produced by Paecilomyces Sp. and Aspergillus Clavatus Isolated from Taxus Mairei and Torreya Grandis
FEMS Immunology and Medical Microbiology 34 (2002) 51^57 www.fems-microbiology.org Brefeldin A, a cytotoxin produced by Paecilomyces sp. and Aspergillus clavatus isolated from Taxus mairei and Torreya grandis Jianfeng Wang a;b;Ã, Yaojian Huang a, Meijuan Fang b, Yongjie Zhang a, Zhonghui Zheng a, Yufen Zhao b, Wenjin Su a;c a The Key Laboratory of Ministry of Education for Cell Biology and Tumor Cell Engineering, School of Life Sciences, Xiamen University, P.O. Box 958, Xiamen 361005, PR China b Bioorganic Phosphorus Chemistry Lab., Department of Chemistry, Xiamen University, Xiamen 361005, PR China c School of Biotechnology, Jimei University, Xiamen 361021, PR China Received 24 April 2002; received in revised form 29 May 2002; accepted 30 May 2002 First published online 3 August 2002 Abstract Paecilomyces sp. and Aspergillus clavatus, which were isolated from Taxus mairei and Torreya grandis from southeast China, produced toxic metabolites when grown in liquid culture. Nuclear magnetic resonance techniques, infrared spectrometry, electrospray ionization mass spectroscopy and X-ray analysis identified brefeldin A, a bioactive metabolite produced by a number of fungal species belonging to the genera Alternaria, Ascochyta, Penicillium, Curvularia, Cercospora and Phyllosticta. This is the first report of the isolation of the cytotoxin from Paecilomyces sp. and A. clavatus. The relevance of brefeldin A to the association between these fungi and their host plants is discussed. ß 2002 Federation of European Microbiological Societies. Published by Elsevier Science B.V. All rights reserved. Keywords: Brefeldin A; Endophytic fungi; Paecilomyces sp.; Aspergillus clavatus; Taxus mairei; Torreya grandis 1. Introduction showed antitumor and antifungal activity, respectively, demonstrating that endophytic fungi are to be a promising Fungi are fundamental to the health and prosperity of source of bene¢cial bioactive products [5]. -

Pharmacogenomics of Ventricular Conduction in Multi-Ethnic Populations Amanda Seyerle Dissertation Committee
Pharmacogenomics of Ventricular Conduction in Multi-Ethnic Populations Amanda Seyerle Dissertation Committee: Christy Avery (chair) Kari North Eric Whitsel Til Stürmer Craig Lee 1 TABLE OF CONTENTS Page LIST OF TABLES ...................................................................................................................................... v LIST OF FIGURES ..................................................................................................................................... vi LIST OF ABBREVIATIONS ..................................................................................................................... vii LIST OF GENE NAMES ............................................................................................................................ ix 1. Overview .............................................................................................................................................. 1 2. Specific Aims ....................................................................................................................................... 3 3. Background and Significance ............................................................................................................ 4 A. Ventricular Conduction ......................................................................................................................... 4 A.1. Electrical Conduction of the Heart ................................................................................................ 4 A.1.1. Sodium Channels.................................................................................................................. -

Brefeldin a | CAS 20350-15-6 | GEF Inhibitor
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic Brefeldin A GEF inhibitor Catalog No. SIH-225 Discovery through partnership | Excellence through quality Overview Product Name Brefeldin A Description GEF inhibitor Purity >98% CAS No. 20350-15-6 Molecular Formula C H O Molecular Weight 280.36 Properties Storage Temperature -20ºC Shipping Temperature Shipped Ambient Product Type Inhibitor Solubility Soluble to 10 mM in ethanol and to 50 mM in DMSO Source Synthetic Appearance White Crystalline Solid SMILES C[C@H]1CCC/C=C/[C@@H]2C[C@@H](C[C@H]2[C@@H](/C=C/C(=O)O1)O)O InChI InChI=1S/C16H24O4/c1-11-5-3-2-4-6-12-9-13(17)10-14(12)15(18)7-8-16(19)20-11/h4,6-8,11-15,17-18H,2-3,5,9-10H2,1H3/b6-4+, InChIKey KQNZDYYTLMIZCT-PNFJWZTBSA-N Safety Phrases Classification: Harmful. May be harmful if inhaled, swallowed or absorbed through skin. Safety Phrases: S22 - Do not breathe dust S24/25 - Avoid contact with skin and eyes S36/37/39 - Wear suitable protective clothing, gloves and eye/face protection Risk Phrases: R68- Possible risk of irreversible effects Hazard Phrases: H301 Precautionary Phrases: P301 + P310 Cite This Product Brefeldin A (StressMarq Biosciences Inc., Victoria BC CANADA, Catalog # SIH-225) Biological Description Alternative Names Cyanaein Research Areas Cancer, Apoptosis PubChem ID 5287620 Scientific Background Brefeldin A is a reversible inhibitor of proteins from the endoplasmic reticulum (ER) to the golgi. -

Unfolded Protein Response and Cell Death After Depletion of Brefeldin A-Inhibited Guanine Nucleotide-Exchange Protein GBF1
Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF1 Carmen Citterio*, Alessandro Vichi*, Gustavo Pacheco-Rodriguez*, Angel M. Aponte†, Joel Moss*, and Martha Vaughan*‡ *Translational Medicine Branch and †Proteomics Core Facility, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892 Contributed by Martha Vaughan, December 31, 2007 (sent for review December 4, 2007) Guanine nucleotide-exchange factors (GEFs) activate ADP- with both class II ARFs. GBF1, which promoted COPI recruit- ribosylation factor (ARF) GTPases that recruit coat proteins to ment to membranes both in vitro and in vivo (12, 13), was membranes to initiate transport vesicle formation. Three mamma- dynamically associated with elements termed vesicular tubular lian GEFs are inhibited by brefeldin A (BFA). GBF1, predominantly clusters (VTCs) and cis-Golgi membranes (14, 15). The GBF1 associated with cis-Golgi membranes, functions early in the secre- molecule contains a dimerization motif, first identified in its tory pathway, whereas BIG1 and BIG2 act in trans-Golgi or later plant homolog GNOM (16), and likely exists as homodimer. sites. Perturbation of endoplasmic reticulum (ER) functions can GBF1 is the BFA-sensitive ARF GEF responsible for membrane result in accumulation of unfolded or misfolded proteins that association of COPI early in the secretory pathway (17), where causes ER stress and unfolded protein response (UPR), with accu- it is essential to the translocation of pre-Golgi intermediates and mulation of ER stress response element (ERSE) gene products. BFA the maintenance of Golgi integrity (18). treatment of cells causes accumulation of proteins in the ER, ER The complex system of ER membranes is the site of synthesis stress, and ultimately apoptosis. -

SAFETY DATA SHEET Revision Date 17.09.2019 According to Regulation (EC) No
Version 6.3 SAFETY DATA SHEET Revision Date 17.09.2019 according to Regulation (EC) No. 1907/2006 Print Date 29.09.2021 GENERIC EU MSDS - NO COUNTRY SPECIFIC DATA - NO OEL DATA SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Brefeldin A Product Number : B7651 Brand : Sigma REACH No. : A registration number is not available for this substance as the substance or its uses are exempted from registration, the annual tonnage does not require a registration or the registration is envisaged for a later registration deadline. CAS-No. : 20350-15-6 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Manufacture of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone number Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 Acute toxicity, Oral (Category 3), H301 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 Label elements Labelling according Regulation (EC) No 1272/2008 Pictogram Signal word Danger Sigma- B7651 Page 1 of 8 The life science business of Merck operates as MilliporeSigma in the US and Canada Hazard statement(s) H301 Toxic if swallowed. -

Gwas Meta-Analysis of Intelligence 1
bioRxiv preprint doi: https://doi.org/10.1101/184853; this version posted September 6, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. GWAS META-ANALYSIS OF INTELLIGENCE 1 GWAS meta-analysis (N=279,930) identifies new genes and functional links to intelligence Jeanne E Savage1#, Philip R Jansen1,2#, Sven Stringer1, Kyoko Watanabe1, Julien Bryois3, Christiaan A de Leeuw1, Mats Nagel1, Swapnil Awasthi4, Peter B Barr5, Jonathan R I Coleman6,7, Katrina L Grasby8, Anke R Hammerschlag1, Jakob Kaminski4,9, Robert Karlsson3, Eva Krapohl6, Max Lam10, Marianne Nygaard11,12, Chandra A Reynolds13, Joey W Trampush14, Hannah Young15, Delilah Zabaneh6, Sara Hägg3, Narelle K Hansell16, Ida K Karlsson3, Sten Linnarsson17, Grant W Montgomery8,18, Ana B Muñoz-Manchado17, Erin B Quinlan6, Gunter Schumann6, Nathan Skene17, Bradley T Webb19,20,21, Tonya White2, Dan E Arking22, Deborah K Attix23,24, Dimitrios Avramopoulos22,25, Robert M Bilder26, Panos Bitsios27, Katherine E Burdick28,29,30, Tyrone D Cannon31, Ornit Chiba-Falek32, Andrea Christoforou23, Elizabeth T Cirulli33, Eliza Congdon26, Aiden Corvin34, Gail Davies35, 36, Ian J Deary35,36, Pamela DeRosse37, Dwight Dickinson38, Srdjan Djurovic39,40, Gary Donohoe41, Emily Drabant Conley42, Johan G Eriksson43,44, Thomas Espeseth45,46, Nelson A Freimer26, Stella Giakoumaki47, Ina Giegling48, Michael Gill34, David