ENZYMATIC MODIFICATION of BACTERIAL EXOPOLYSACCHARIDES Xanthan Lyase As Atoo L for Structural Andfunctiona L Modification of Xanthan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Beno Cornellgrad 0058F 10677.Pdf
BACILLALES INFLUENCE QUALITY AND SAFETY OF DAIRY PRODUCTS A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Sarah Marie Beno December 2017 © 2017 Sarah Marie Beno BACILLALES INFLUENCE QUALITY AND SAFETY OF DAIRY PRODUCTS Sarah Marie Beno, Ph. D. Cornell University 2017 Bacillales, an order of Gram-positive bacteria, are commonly isolated from dairy foods and at various points along the dairy value chain. Three families of Bacillales are analyzed in this work: (i) Listeriaceae (represented by Listeria monocytogenes), (ii) Paenibacillaceae (represented by Paenibacillus), and (iii) Bacillaceae (represented by the Bacillus cereus group). These families impact both food safety and food quality. Most Listeriaceae are non- pathogenic, but L. monocytogenes has one of the highest mortality rates of foodborne pathogens. Listeria spp. are often reported in food processing environments. Here, 4,430 environmental samples were collected from 9 small cheese-processing facilities and tested for Listeria and L. monocytogenes. Prevalence varied by processing facility, but across all facilities, 6.03 and 1.35% of samples were positive for L. monocytogenes and other Listeria spp., respectively. Each of these families contains strains capable of growth at refrigeration temperatures. To more broadly understand milk spoilage bacteria, genetic analyses were performed on 28 Paenibacillus and 23 B. cereus group isolates. While no specific genes were significantly associated with cold-growing Paenibacillus, the growth variation and vast genetic data introduced in this study provide a strong foundation for the development of detection strategies. Some species within the B. -

X-Ray Fluorescence Analysis Method Röntgenfluoreszenz-Analyseverfahren Procédé D’Analyse Par Rayons X Fluorescents
(19) & (11) EP 2 084 519 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: G01N 23/223 (2006.01) G01T 1/36 (2006.01) 01.08.2012 Bulletin 2012/31 C12Q 1/00 (2006.01) (21) Application number: 07874491.9 (86) International application number: PCT/US2007/021888 (22) Date of filing: 10.10.2007 (87) International publication number: WO 2008/127291 (23.10.2008 Gazette 2008/43) (54) X-RAY FLUORESCENCE ANALYSIS METHOD RÖNTGENFLUORESZENZ-ANALYSEVERFAHREN PROCÉDÉ D’ANALYSE PAR RAYONS X FLUORESCENTS (84) Designated Contracting States: • BURRELL, Anthony, K. AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Los Alamos, NM 87544 (US) HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE SI SK TR (74) Representative: Albrecht, Thomas Kraus & Weisert (30) Priority: 10.10.2006 US 850594 P Patent- und Rechtsanwälte Thomas-Wimmer-Ring 15 (43) Date of publication of application: 80539 München (DE) 05.08.2009 Bulletin 2009/32 (56) References cited: (60) Divisional application: JP-A- 2001 289 802 US-A1- 2003 027 129 12164870.3 US-A1- 2003 027 129 US-A1- 2004 004 183 US-A1- 2004 017 884 US-A1- 2004 017 884 (73) Proprietors: US-A1- 2004 093 526 US-A1- 2004 235 059 • Los Alamos National Security, LLC US-A1- 2004 235 059 US-A1- 2005 011 818 Los Alamos, NM 87545 (US) US-A1- 2005 011 818 US-B1- 6 329 209 • Caldera Pharmaceuticals, INC. US-B2- 6 719 147 Los Alamos, NM 87544 (US) • GOLDIN E M ET AL: "Quantitation of antibody (72) Inventors: binding to cell surface antigens by X-ray • BIRNBAUM, Eva, R. -
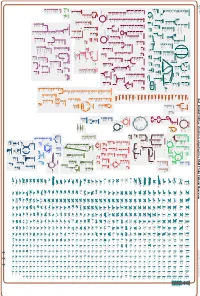
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000263195Cyc: Emticicia oligotrophica DSM 17448 Cellular Overview Connections between pathways are omitted for legibility. -

Xanth-On-Template-10May Clean
This is a repository copy of Structural dynamics and catalytic properties of a multimodular xanthanase. White Rose Research Online URL for this paper: https://eprints.whiterose.ac.uk/132388/ Version: Accepted Version Article: Moroz, Olga V., Jensen, Pernille F., McDonald, Sean P. et al. (17 more authors) (2018) Structural dynamics and catalytic properties of a multimodular xanthanase. ACS Catalysis. 6021–6034. ISSN 2155-5435 https://doi.org/10.1021/acscatal.8b00666 Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Structural Dynamics and Catalytic Properties of a Multi-Modular Xanthanase Olga V. Moroz1,=, Pernille F. Jensen2,=, Sean P. McDonald3,=, Nicholas McGregor4, Elena Blagova1, Gerard Comamala2, Dorotea R. Segura5, Lars Anderson5, Santhosh M. Vasu5, Vasudeva P. Rao5, Lars Giger5, Trine Holst Sørensen6, Rune Nygaard Monrad5, Allan Svendsen5, Jens E. Nielsen5, Bernard -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

A Hierarchical Classification of Polysaccharide Lyases for Glycogenomics Vincent Lombard, Thomas Bernard, Corinne Rancurel, Harry Brumer, Pedro M
A hierarchical classification of polysaccharide lyases for glycogenomics Vincent Lombard, Thomas Bernard, Corinne Rancurel, Harry Brumer, Pedro M. Coutinho, Bernard Henrissat To cite this version: Vincent Lombard, Thomas Bernard, Corinne Rancurel, Harry Brumer, Pedro M. Coutinho, et al.. A hierarchical classification of polysaccharide lyases for glycogenomics. Biochemical Journal, Portland Press, 2010, 432 (3), pp.437-444. 10.1042/BJ20101185. hal-00539724 HAL Id: hal-00539724 https://hal.archives-ouvertes.fr/hal-00539724 Submitted on 25 Nov 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 07 Oct 2010 as manuscript BJ20101185 A hierarchical classification of polysaccharide lyases for glycogenomics V. Lombard*, T. Bernard*†, C. Rancurel*, H Brumer‡, P.M. Coutinho* & B. Henrissat*1 *Architecture et Fonction des Macromolécules Biologiques, UMR6098, CNRS, Université de la Méditerranée, Université de Provence, Case 932, 163 Avenue de Luminy, 13288 Marseille cedex 9, France ‡School of Biotechnology, Royal Institute of Technology (KTH), AlbaNova University Centre, 106 91 Stockholm, Sweden † Present address: Biométrie et Biologie Évolutive, UMR CNRS 5558, UCB Lyon 1, Bât. Grégor Mendel, 43 bd du 11 novembre 1918, 69622 Villeurbanne cedex, France 1To whom correspondence should be addressed: [email protected]‐mrs.fr Abstract: Carbohydrate‐active enzymes face huge substrate diversity in a highly selective manner with only a limited number of available folds. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0132824 A1 Segura Et Al
US 2015O132824A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0132824 A1 Segura et al. (43) Pub. Date: May 14, 2015 (54) POLYPEPTIDES HAVING XANTHAN (30) Foreign Application Priority Data DEGRADING ACTIVITY AND POLYNUCLEOTDESENCODING SAME May 7, 2012 (EP) .................................. 12167O23.6 Jan. 10, 2013 (EP) .................................. 13150833.5 (71) Applicant: Novozymes A/S, Bagsvaerd (DK) (72) Inventors: Dorotea Raventos Segura, Rungsted Publication Classification (DK); Peter Fischer Halin, Holte (DK); Anders Viksoe-Nielsen, Slangerup (51) Int. C. (DK); Lars Anderson, Malmoe (SE): CI2N 9/42 (2006.01) Martin Simon Borchert, Hilleroed (DK); Leigh Murphy, Roskilde (DK); CID 3/386 (2006.01) Astrid Boisen, Soeborg (DK); Lorena C09K8/08 (2006.01) G. Palmén, Malmoe (SE); Kenneth CI2N 9/88 (2006.01) Jensen, Oelsted (DK); Carsten (52) U.S. C. Sjoeholm, Virum (DK); Tine Hoff, CPC ................ CI2N 9/2437 (2013.01); C12N 9/88 Holte (DK); Charlotte Blom, Lynge (2013.01): CII D3/38645 (2013.01): CIID (DK) 3/386.36 (2013.01); C09K8/08 (2013.01); C12Y 302/01004 (2013.01); C12Y402/02012 (21) Appl. No.: 14/395,165 (2013.01) (22) PCT Fled: May 7, 2013 (57) ABSTRACT (86) PCT NO.: PCT/EP2013/059472 S371 (c)(1), The present invention relates to isolated polypeptides having (2) Date: Oct. 17, 2014 Xanthan degrading activity, catalytic domains and polynucle otides encoding the polypeptides and catalytic domains. The Related U.S. Application Data invention also relates to nucleic acid constructs, vectors, and (60) Provisional application No. 61/644,033, filed on May host cells comprising the polynucleotides as well as methods 8, 2012, provisional application No. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Enzymatic Modification and Characterization of Xanthan
Enzymatic modification and characterization of xanthan Marijn M. Kool Thesis committee Promotor Prof. Dr. Ir. H. Gruppen Professor of Food Chemistry Wageningen University Co‐promotor Prof. Dr H.A. Schols Personal chair at the Laboratory of Food Chemistry Wageningen University Other members Dr T.J. Foster, University of Nottingham, United Kingdom Prof. Dr J.G.M Janssen, University of Amsterdam, The Netherlands Prof. Dr J. van der Oost, Wageningen University, The Netherlands Prof. Dr C. Sandström, Swedish University of Agricultural Sciences, Uppsala, Sweden This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Enzymatic modification and characterization of xanthan Marijn M. Kool Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff. in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 7 February 2014 at 04.00 p.m. in the Aula. Marijn M. Kool Enzymatic modification and characterization of xanthan 144 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in English and Dutch ISBN: 978‐94‐6173‐865‐3 ABSTRACT In this thesis an enzymatic approach for the modification and characterization of xanthans was introduced. Complete backbone degradation of xanthan by cellulases was obtained independent on the molar composition of a xanthan sample. It was shown that only xanthan segments that occurred in a disordered xanthan conformation were susceptible to enzymatic backbone degradation. HILIC‐ELSD‐MS analysis revealed the presence of six different xanthan repeating units (RUs). -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -
Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. E3m18Cyc: Arenibacter sp. E3M18 Cellular Overview Connections between pathways are omitted for legibility. a siderophore 1-(β-D Fe 3+ cefoperazone a siderophore a siderophore Fe 3+ Fe 3+ ribofuranosyl) glucosyl-oleandomycin N-acetyl-α-D- 3+ 3+ N-acetyl-α-D- N-acetyl-α-D- Fe Fe nicotinamide glucosamine glucosamine glucosamine Ferric Ferric Ferric siderophore ABC- iron(III) ABC Ribosyl Multidrug N-acetylglucosamine N-acetylglucosamine N-acetylglucosamine siderophore siderophore transport type Fe3+ transporter, nicotinamide resistance related transporter, related related transporter, transport transport system, transport ATP-binding transporter, protein transporter, NagX system, system, periplasmic system protein, binding protein; putative protein TonB cefoperazone N-acetyl-α-D- N-acetyl-α-D- β N-acetyl-α-D- 1-( -D glucosyl-oleandomycin glucosamine glucosamine ribofuranosyl) glucosamine a siderophore a siderophore Fe 3+ Fe 3+ Fe 3+ Fe 3+ nicotinamide a siderophore Fe 3+ tolB protein Amino Acid Degradation Cofactor, Prosthetic Group, Electron Carrier, and Vitamin Biosynthesis Aromatic Compound -

Characterization of a Hyaluronic Acid Utilization Locus and Identification of Two Hyaluronate Lyases in a Marine Bacterium Vibrio Alginolyticus LWW-9
ORIGINAL RESEARCH published: 10 June 2021 doi: 10.3389/fmicb.2021.696096 Characterization of a Hyaluronic Acid Utilization Locus and Identification of Two Hyaluronate Lyases in a Marine Bacterium Vibrio alginolyticus LWW-9 Xiaoyi Wang 1,2, Ziwei Wei 1,2, Hao Wu 1,2, Yujiao Li 1,2, Feng Han 1,2* and Wengong Yu 1,2* 1 Shandong Provincial Key Laboratory of Glycoscience and Glycoengineering, School of Medicine and Pharmacy, Ocean University of China, Qingdao, China, 2 Laboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China Hyaluronic acid (HA) is a negatively charged and linear polysaccharide existing in the tissues and body fluids of all vertebrates. Some pathogenic bacteria target hyaluronic acid for adhesion and/or infection to host cells. Vibrio alginolyticus is an opportunistic pathogen related to infections of humans and marine animals, and the hyaluronic acid- Edited by: degrading potential of Vibrio spp. has been well-demonstrated. However, little is known Benwei Zhu, Nanjing Tech University, China about how Vibrio spp. utilize hyaluronic acid. In this study, a marine bacterium V. Reviewed by: alginolyticus LWW-9 capable of degrading hyaluronic acid has been isolated. Genetic Damao Wang, and bioinformatic analysis showed that V. alginolyticus LWW-9 harbors a gene cluster Southwest University, China Yayue Wang, involved in the degradation, transport, and metabolism of hyaluronic acid. Two novel PL8 Shangqiu Normal University, China family hyaluronate lyases, VaHly8A and VaHly8B, are the key enzymes for the degradation *Correspondence: of hyaluronic acid. VaHly8A and VaHly8B have distinct biochemical properties, reflecting Feng Han the adaptation of the strain to the changing parameters of the aquatic habitats and hosts.