Genetic Variants in Epigenetic Pathways and Risks of Multiple Cancers in the GAME-ON Consortium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rabbit Anti-SEN1/FITC Conjugated Antibody
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-SEN1/FITC Conjugated antibody SL7576R-FITC Product Name: Anti-SEN1/FITC Chinese Name: FITC标记的细胞衰老相关蛋白1抗体 Cell senescence related gene complementation group B; Cellular senescence related protein 1; CSR; CSRB; MORF 4; MORF4; MORF4 protein; Mortality factor 4; SEN 1; Alias: SEN; Senescence (cellular) related 1; Senescence related cellular 1; Transcription factor like protein MORF4; MO4L1_HUMAN; Mortality factor 4-like protein 1; MORF- related gene 15 protein; Protein MSL3-1; Transcription factor-like protein MRG15. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Cow,Zebrafish, IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 27kDa Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human Mortality factor 4-like protein 1 Lsotype: IgGwww.sunlongbiotech.com Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: Cellular senescence, the terminal nondividing state that normal cells enter following completion of their proliferative potential, is the dominant phenotype in hybrids of Product Detail: normal and immortal cells. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Research article Open Access The functional modulation of epigenetic regulators by alternative splicing Sergio Lois1,2, Noemí Blanco1,2, Marian Martínez-Balbás*1,2 and Xavier de la Cruz*2,3 Address: 1Instituto de Biología Molecular de Barcelona. CID. Consejo Superior de Investigaciones Científicas (CSIC); 08028 Barcelona, Spain, 2Institut de Recerca Biomèdica-PCB; 08028 Barcelona, Spain and 3Institució Catalana de Recerca i Estudis Avançats (ICREA); Barcelona, Spain Email: Sergio Lois - [email protected]; Noemí Blanco - [email protected]; Marian Martínez-Balbás* - [email protected]; Xavier de la Cruz* - [email protected] * Corresponding authors Published: 25 July 2007 Received: 24 April 2007 Accepted: 25 July 2007 BMC Genomics 2007, 8:252 doi:10.1186/1471-2164-8-252 This article is available from: http://www.biomedcentral.com/1471-2164/8/252 © 2007 Lois et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Epigenetic regulators (histone acetyltransferases, methyltransferases, chromatin- remodelling enzymes, etc) play a fundamental role in the control of gene expression by modifying the local state of chromatin. However, due to their recent discovery, little is yet known about their own regulation. This paper addresses this point, focusing on alternative splicing regulation, a mechanism already known to play an important role in other protein families, e.g. transcription factors, membrane receptors, etc. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Understanding the Role of the Chromosome 15Q25.1 in COPD Through Epigenetics and Transcriptomics
European Journal of Human Genetics (2018) 26:709–722 https://doi.org/10.1038/s41431-017-0089-8 ARTICLE Understanding the role of the chromosome 15q25.1 in COPD through epigenetics and transcriptomics 1 1,2 1,3,4 5,6 5,6 Ivana Nedeljkovic ● Elena Carnero-Montoro ● Lies Lahousse ● Diana A. van der Plaat ● Kim de Jong ● 5,6 5,7 6 8 9 10 Judith M. Vonk ● Cleo C. van Diemen ● Alen Faiz ● Maarten van den Berge ● Ma’en Obeidat ● Yohan Bossé ● 11 1,12 12 1 David C. Nickle ● BIOS Consortium ● Andre G. Uitterlinden ● Joyce J. B. van Meurs ● Bruno C. H. Stricker ● 1,4,13 6,8 5,6 1 1 Guy G. Brusselle ● Dirkje S. Postma ● H. Marike Boezen ● Cornelia M. van Duijn ● Najaf Amin Received: 14 March 2017 / Revised: 6 November 2017 / Accepted: 19 December 2017 / Published online: 8 February 2018 © The Author(s) 2018. This article is published with open access Abstract Chronic obstructive pulmonary disease (COPD) is a major health burden in adults and cigarette smoking is considered the most important environmental risk factor of COPD. Chromosome 15q25.1 locus is associated with both COPD and smoking. Our study aims at understanding the mechanism underlying the association of chromosome 15q25.1 with COPD through epigenetic and transcriptional variation in a population-based setting. To assess if COPD-associated variants in 1234567890();,: 15q25.1 are methylation quantitative trait loci, epigenome-wide association analysis of four genetic variants, previously associated with COPD (P < 5 × 10−8) in the 15q25.1 locus (rs12914385:C>T-CHRNA3, rs8034191:T>C-HYKK, rs13180: C>T-IREB2 and rs8042238:C>T-IREB2), was performed in the Rotterdam study (n = 1489). -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Functional Mechanisms Underlying Pleiotropic Risk Alleles at the 19P13.1 Breast&Ndash;Ovarian Cancer Susceptibility Locus
ARTICLE Received 16 Jun 2015 | Accepted 20 Jul 2016 | Published 7 Sep 2016 DOI: 10.1038/ncomms12675 OPEN Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast–ovarian cancer susceptibility locus Kate Lawrenson et al.# A locus at 19p13 is associated with breast cancer (BC) and ovarian cancer (OC) risk. Here we analyse 438 SNPs in this region in 46,451 BC and 15,438 OC cases, 15,252 BRCA1 mutation carriers and 73,444 controls and identify 13 candidate causal SNPs associated with serous OC (P ¼ 9.2 Â 10 À 20), ER-negative BC (P ¼ 1.1 Â 10 À 13), BRCA1-associated BC (P ¼ 7.7 Â 10 À 16) and triple negative BC (P-diff ¼ 2 Â 10 À 5). Genotype-gene expression associations are identified for candidate target genes ANKLE1 (P ¼ 2 Â 10 À 3) and ABHD8 (Po2 Â 10 À 3). Chromosome conformation capture identifies interactions between four candidate SNPs and ABHD8, and luciferase assays indicate six risk alleles increased transactivation of the ADHD8 promoter. Targeted deletion of a region containing risk SNP rs56069439 in a putative enhancer induces ANKLE1 downregulation; and mRNA stability assays indicate functional effects for an ANKLE1 30-UTR SNP. Altogether, these data suggest that multiple SNPs at 19p13 regulate ABHD8 and perhaps ANKLE1 expression, and indicate common mechanisms underlying breast and ovarian cancer risk. Correspondence and requests for materials should be addressed to A.C.A. (email: [email protected]). #A full list of authors and their affiliations appears at the end of the paper. -
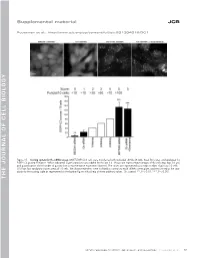
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Functional Mechanisms Underlying Pleiotropic Risk Alleles at the 19P13.1 Breast&Ndash;Ovarian Cancer Susceptibility Locus
UC Office of the President Recent Work Title Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast–ovarian cancer susceptibility locus Permalink https://escholarship.org/uc/item/1rx2f44n Author Lawrenson, Kate Publication Date 2016-09-07 DOI 10.1038/ncomms12675 Peer reviewed eScholarship.org Powered by the California Digital Library University of California ARTICLE Received 16 Jun 2015 | Accepted 20 Jul 2016 | Published 7 Sep 2016 DOI: 10.1038/ncomms12675 OPEN Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast–ovarian cancer susceptibility locus Kate Lawrenson et al.# A locus at 19p13 is associated with breast cancer (BC) and ovarian cancer (OC) risk. Here we analyse 438 SNPs in this region in 46,451 BC and 15,438 OC cases, 15,252 BRCA1 mutation carriers and 73,444 controls and identify 13 candidate causal SNPs associated with serous OC (P ¼ 9.2 Â 10 À 20), ER-negative BC (P ¼ 1.1 Â 10 À 13), BRCA1-associated BC (P ¼ 7.7 Â 10 À 16) and triple negative BC (P-diff ¼ 2 Â 10 À 5). Genotype-gene expression associations are identified for candidate target genes ANKLE1 (P ¼ 2 Â 10 À 3) and ABHD8 (Po2 Â 10 À 3). Chromosome conformation capture identifies interactions between four candidate SNPs and ABHD8, and luciferase assays indicate six risk alleles increased transactivation of the ADHD8 promoter. Targeted deletion of a region containing risk SNP rs56069439 in a putative enhancer induces ANKLE1 downregulation; and mRNA stability assays indicate functional effects for an ANKLE1 30-UTR SNP. Altogether, these data suggest that multiple SNPs at 19p13 regulate ABHD8 and perhaps ANKLE1 expression, and indicate common mechanisms underlying breast and ovarian cancer risk. -

SPINT2 Suppresses Hippo Effector YAP and Limits Cellular Tolerance for Aneuploidy
SPINT2 Suppresses Hippo Effector YAP and Limits Cellular Tolerance for Aneuploidy The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Zhang, Huadi. 2017. SPINT2 Suppresses Hippo Effector YAP and Limits Cellular Tolerance for Aneuploidy. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42061511 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA SPINT2 Suppresses Hippo Effector YAP and Limits Cellular Tolerance for Aneuploidy A dissertation presented by Huadi Zhang to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biological and Biomedical Sciences Harvard University Cambridge, Massachusetts August 2017 © 2017 Huadi Zhang All rights reserved. Dissertation Advisor: Professor David Pellman Huadi Zhang SPINT2 Suppresses Hippo Effector YAP and Limits Cellular Tolerance for Aneuploidy Abstract Oncogenic transformation is often accompanied by chromosome instability, an increased rate of chromosome missegregation. The consequent gain or loss of chromosomes—termed aneuploidy—hinders the growth of most non-cancerous tissues, but is prevalent in tumors. During tumorigenesis, aneuploidy contributes to cellular heterogeneity and may promote downstream mutations, including chromosome rearrangements and oncogene amplification. Cellular mechanisms that safeguard against aneuploidy remain unclear. The Hippo pathway is a tumor-suppressor mechanism with essential roles in regulating tissue homeostasis. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Supplemental Material 1
SUP. FIGURE S1 DAPI NFAT Merge DMSO Ac5SGlcNAc Figure S1. Inhibition of OGT does not prevent nuclear translocation of NFAT. Jurkat cells !"#$%&'()*+(!!,-.'/012"#..(3'4056'7(+('"+(#"(3'7,"8'9:';<'5=29>/%=45='?+'@<>A'B?+'CD' 8+!E''F(%%!'7(+('"8(-'*%#"(3'?-'#-",2F@GHF@ID2=?#"(3'=?J(+!%,*!'B?+'G:'K,-!E''5B"(+'B,)#",?-L' =(%%!'7(+('#-#%&M(3'$&'=?-B?=#%'K,=+?!=?*&E SUP. FIGURE S2 Labeled Unlabeled Labeled Unlabeled 1h 18h Az PEG 1h 19h Az PEG !"#$%"#&'( + + + + !"#$%"#&'( + + + + 110 EWSR1 160 *34+&5 80 &,/ 160 )*+-2$ 160 110 )*+,& 80 60 &,/ 160 110 80 0"1"- RUNX1 110 80 60 60 50 160 SP1 160 110 ELF1 110 80 160 NUP98 110 80 )*+&-. &,/ Figure S2. PEG mass tags allow visualization of OGlcNAc stoichiometry. 5K PEG mass tags were affixed to OGlcNAc groups on proteins from control or activated T cells via enzy matic labeling with azide and copperfree click chemistry. Proteins were then analyzed for shifts in electrophoretic mobility by immunoblot. For the unlabeled control samples, either the azide (Az) or PEG (PEG) reagent was omitted during the labeling procedure. Note that HCFC1 appears as multiple bands because the protein is expressed as a single polypeptide that under goes proteolytic processing. UBAP2L appears as two bands in the unlabeled control samples due to alternative splicing. Table S1. Details of 133 higher confidence and 81 lower confidence O-GlcNAc glycoproteins1. Confidence Uniprot ID Symbol Specificity Chi Higher P55265 ADAR 100% 9.11E-04 Higher Q09666 AHNAK 92% 2.36E-16 Higher Q8IWZ3 ANKHD1 100% 1.19E-03 Higher