An Introduction to Genetics for Mathematicians Emily Scheele
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Introduction to Genetics Bios 225
INTRODUCTION TO GENETICS BIOS 225 Course Description This course introduces the student to the basic concepts of inheritance, populations, mutations, and techniques used to assess each of these. Credit: 2 credits Repeatable: No Course Structure The course will be presented in different formats: Lectures with PowerPoints, self-directed learning, discussions and student assignments etc. Competencies This course emphasizes competencies to enhance skills essential for a future health care professional. • Knowledge o Demonstrate content knowledge and skills in foundational courses required by biomedical professionals o Demonstrate information literacy o Demonstrate quantitative reasoning o Demonstrate longitudinal learning through coursework • Critical Thinking o Develop the skills of self-reflection and peer assessment to improve personal performance. o Demonstrate the ability to analyze literature and written material o Demonstrate the ability to distinguish between well-reasoned and poorly reasoned arguments • Communication Skills o Demonstrate effective presentation skills to faculty and peers. o Demonstrate effective listening skills o Demonstrate effective written communication 1 Objectives: Upon completion of BIOS 225 course, the student should be able to describe: • The structure and function of purines, pyrimidines, nucleosides and nucleotides • The structure and functions of nucleic acids (DNA and RNA) • The chromosome anatomy and human karyotypes • The concepts of prokaryotic and eukaryotic DNA replication • The concepts of prokaryotic and eukaryotic RNA transcription and post-transcriptional modifications • The concepts of prokaryotic and eukaryotic protein translation and post-translational modifications • The regulations of prokaryotic and eukaryotic gene expressions • The process of genomic, chromosomal and gene mutations; and its repair mechanism • The Mendel’ hypothesis and molecular mechanisms of genetic inheritance Schedule: Dates and times to be posted at the beginning of the term on the online calendar. -

Animal Traditions: Behavioural Inheritance in Evolution
Animal Traditions: Behavioural Inheritance in Evolution Eytan Avital and Eva Jablonka CAMBRIDGE UNIVERSITY PRESS Animal Traditions Behavioural Inheritance in Evolution Animal Traditions maintains that the assumption that the selection of genes supplies both a sufficient explanation of the evolution of behav- iour and a true description of its course is, despite its almost univer- sal acclaim, wrong. Eytan Avital and Eva Jablonka contend that evolutionary explanations must take into account the well-established fact that, in mammals and birds, the transfer of learnt information across generations is both ubiquitous and indispensable. The introduc- tion of the behavioural inheritance system into the Darwinian explanatory scheme enables the authors to offer new interpretations for common behaviours such as maternal behaviours, behavioural conflicts within families, adoption and helping. This approach offers a richer view of heredity and evolution, integrates developmental and evolutionary processes, suggests new lines for research and provides a constructive alternative to both the selfish gene and meme views of the world. It will make stimulating reading for all those interested in evolutionary biology, sociobiology, behavioural ecology and psychology. eytan avital is a lecturer in Zoology in the Department of Natural Sciences at David Yellin College of Education in Jerusalem. He is a highly experienced field biologist, and has written one zoology text and edited several others on zoology and evolution for the Israel Open university. eva jablonka is a senior lecturer in the Cohn Institute for the History and Philosophy of Science and Ideas, at Tel-Aiv University. She is the author of three books on heredity and evolution, most recently Epigenetic Inheritance and Evolution with Marion Lamb. -
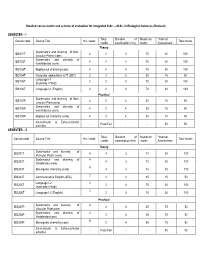
Detailed Course Matrix and Scheme of Evaluation for Integrated B.Sc. – M.Sc. in Biological Sciences (Revised) SEMESTER –
Detailed course matrix and scheme of evaluation for Integrated B.Sc. – M.Sc. in Biological Sciences (Revised) SEMESTER – I Total Duration of Maximum *Internal Course code Course Title Hrs / week Total marks credits examination (hrs) marks Assessment Theory Systematics and diversity of Non- IBS101T 4 4 3 70 30 100 vascular Plants (core) Systematics and diversity of IBS102T 4 4 3 70 30 100 Invertebrates (core) IBS103T Biophysical chemistry(core) 4 4 3 70 30 100 IBS104T Computer applications & IT (SEC) 2 2 2 35 15 50 Language I-1 IBS105T 2 2 3 70 30 100 (Kannada / Hindi) IBS106T Language I-1 (English) 2 2 3 70 30 100 Practical Systematics and diversity of Non- IBS107P 4 2 4 35 15 50 vascular Plant (core) Systematics and diversity of IBS108P 4 2 4 35 15 50 Invertebrates (core) IBS109P Biophysical chemistry (core) 4 2 4 35 15 50 Co-curricular & Extra-curricular - Pass/Fail - - 50 50 activities SEMESTER – II Total Duration of Maximum *Internal Course code Course Title Hrs / week Total marks credits examination (hrs) marks Assessment Theory Systematics and diversity of IBS201T 4 4 3 70 30 100 Vascular Plants (core) Systematics and diversity of 4 IBS202T 4 3 70 30 100 Vertebrates (core) 4 IBS203T Bio-organic chemistry (core) 4 3 70 30 100 2 IBS204T Communicative English (AEC) 2 2 35 15 50 Language I-2 2 IBS205T 2 3 70 30 100 (Kannada / Hindi) 2 IBS206T Language I-2 (English) 2 3 70 30 100 Practical Systematics and diversity of 4 IBS207P 2 4 35 15 50 Vascular Plant(core) Systematics and diversity of 4 IBS208P 2 4 35 15 50 Vertebrates(core) 4 IBS209P -

INTRODUCTION to GENETICS Table of Contents Heredity, Historical
INTRODUCTION TO GENETICS Table of Contents Heredity, historical perspectives | The Monk and his peas | Principle of segregation Dihybrid Crosses | Mutations | Genetic Terms | Links Heredity, Historical Perspective | Back to Top For much of human history people were unaware of the scientific details of how babies were conceived and how heredity worked. Clearly they were conceived, and clearly there was some hereditary connection between parents and children, but the mechanisms were not readily apparent. The Greek philosophers had a variety of ideas: Theophrastus proposed that male flowers caused female flowers to ripen; Hippocrates speculated that "seeds" were produced by various body parts and transmitted to offspring at the time of conception, and Aristotle thought that male and female semen mixed at conception. Aeschylus, in 458 BC, proposed the male as the parent, with the female as a "nurse for the young life sown within her". During the 1700s, Dutch microscopist Anton van Leeuwenhoek (1632-1723) discovered "animalcules" in the sperm of humans and other animals. Some scientists speculated they saw a "little man" (homunculus) inside each sperm. These scientists formed a school of thought known as the "spermists". They contended the only contributions of the female to the next generation were the womb in which the homunculus grew, and prenatal influences of the womb. An opposing school of thought, the ovists, believed that the future human was in the egg, and that sperm merely stimulated the growth of the egg. Ovists thought women carried eggs containing boy and girl children, and that the gender of the offspring was determined well before conception. -

2014-2015 and Is Accurate and Current, to the Greatest Extent Possible, As of June 2014
Cover Cover 1 University’s Mission Statement James B. Duke’s founding Indenture of Duke University directed the members of the University to “provide real leadership in the educational world” by choosing individuals of “outstanding character, ability and vision” to serve as its officers, trustees and faculty; by carefully selecting students of “character, determination and application;” and by pursuing those areas of teaching and scholarship that would “most help to develop our resources, increase our wisdom and promote human happiness.” To these ends, the mission of Duke University is to provide a superior liberal education to undergraduate students, attending not only to their intellectual growth but also to their development as adults committed to high ethical standards and full participation as leaders in their communities; to prepare future members of the learned professions for lives of skilled and ethical service by providing excellent graduate and professional education; to advance the frontiers of knowledge and contribute boldly to the international community of scholarship; to promote an intellectual environment built on a commitment to free and open inquiry; to help those who suffer, cure disease and promote health, through sophisticated medical research and thoughtful patient care; to provide wide ranging educational opportunities, on and beyond our campuses, for traditional students, active professionals and life-long learners using the power of information technologies; and to promote a deep appreciation for the range of human difference and potential, a sense of the obligations and rewards of citizenship, and a commitment to learning, freedom and truth. By pursuing these objectives with vision and integrity, Duke University seeks to engage the mind, elevate the spirit, and stimulate the best effort of all who are associated with the University; to contribute in diverse ways to the local community, the state, the nation and the world; and to attain and maintain a place of real leadership in all that we do. -

Mechanisms and Consequences of Epigenetic Inheritance Following Parental Preconception Alcohol Exposure
Loyola University Chicago Loyola eCommons Dissertations Theses and Dissertations 2018 Mechanisms and Consequences of Epigenetic Inheritance Following Parental Preconception Alcohol Exposure Annadorothea Asimes Loyola University Chicago Follow this and additional works at: https://ecommons.luc.edu/luc_diss Part of the Neurosciences Commons Recommended Citation Asimes, Annadorothea, "Mechanisms and Consequences of Epigenetic Inheritance Following Parental Preconception Alcohol Exposure" (2018). Dissertations. 2770. https://ecommons.luc.edu/luc_diss/2770 This Dissertation is brought to you for free and open access by the Theses and Dissertations at Loyola eCommons. It has been accepted for inclusion in Dissertations by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. Copyright © 2018 Annadorothea Asimes LOYOLA UNIVERSITY CHICAGO MECHANISMS AND CONSEQUENCES OF EPIGENETIC INHERITANCE FOLLOWING PARENTAL PRECONCEPTION ALCOHOL EXPOSURE A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PROGRAM IN NEUROSCIENCE BY ANNADOROTHEA ASIMES CHICAGO, IL MAY 2018 Copyright by AnnaDorothea Asimes, 2018 ALL rights reserved. ACKNOWLEDGEMENTS The work required to compLete this dissertation wouLd not have been possibLe alone. I wouLd first Like to thank my advisor and mentor, Dr. Toni Pak. The guidance and support you have given me throughout these graduate schooL years has been invaluabLe. You have provided a chalLenging environment that pushed me to be resourcefuL, creative, boLd, and humbLed. I wouLd not be the scientist I am today without your mentorship and friendship. I wouLd also Like to thank my Lab mates, who quickLy turned from coworkers to friends. I Look forward to buiLding our Lab famiLy and seeing the great work you alL go on to do. -

Mendelian Gene ” and the ” Molecular Gene ” : Two Relevant Concepts of Genetic Units V Orgogozo, Alexandre E
The ” Mendelian Gene ” and the ” Molecular Gene ” : Two Relevant Concepts of Genetic Units V Orgogozo, Alexandre E. Peluffo, Baptiste Morizot To cite this version: V Orgogozo, Alexandre E. Peluffo, Baptiste Morizot. The ” Mendelian Gene ” and the ”Molec- ular Gene ” : Two Relevant Concepts of Genetic Units. Virginie Orgogozo. Genes and Evolu- tion, 119, Elsevier, pp.1-26, 2016, Current Topics in Developmental Biology, 978-0-12-417194-7. 10.1016/bs.ctdb.2016.03.002. hal-01354346 HAL Id: hal-01354346 https://hal.archives-ouvertes.fr/hal-01354346 Submitted on 19 Aug 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ARTICLE IN PRESS The “Mendelian Gene” and the “Molecular Gene”: Two Relevant Concepts of Genetic Units V. Orgogozo*,1, A.E. Peluffo*, B. Morizot† *Institut Jacques Monod, UMR 7592, CNRS-Universite´ Paris Diderot, Sorbonne Paris Cite´, Paris, France †Universite´ Aix-Marseille, CNRS UMR 7304, Aix-en-Provence, France 1Corresponding author: e-mail address: [email protected] Contents 1. Introduction 2 2. The “Mendelian Gene” and the “Molecular Gene” 5 3. Current Literature Often Confuses the “Mendelian Gene” and the “Molecular Gene” Concepts 7 4. -

BIO 362 – Introduction to Genetics
PSS6325 – Epigenetic Mechanisms (Fall 2017) 1. Instructor Dr. Benildo G. de los Reyes Professor and Bayer CropScience Endowed Chair in Plant Genomics Department of Plant and Soil Science Experimental Sciences Building, Room 215; 806-834-6421; [email protected] Consultation: By appointment 2. Textbook (Recommended but not required; Available at the library on reserve) Allis CD, Caparros ML, Jenuwein T, Reinberg D (2015) Epigenetics, 2nd Edition, Cold Spring Harbor Laboratory Press, ISBN:978-1-936113-59-0 or newer edition if available. Lecture hand-outs and power point slides will be accessible to students in electronic or other forms. Supplementary printed reading materials will also be provided as necessary. 3. Course description Prerequisites: Consent of instructor; 3-credits, lecture Epigenetics is the study of phenotypic expression that cannot be fully explained by Mendelian principles of heredity, and by DNA sequence variation among individuals. The core of this science is rooted from intrinsically and extrinsically determined processes that lead to dynamic changes in chromatin structure, DNA methylation, RNA- directed gene regulation and genome modification, and how these processes bring about heritable phenotypic variation at the somatic and/or germ levels. During the last decade, the study of epigenetic mechanisms has become a critical aspect in understanding how plants and animals develop and adapt to environmental changes and diseases, providing another layer of complexity to the flow of genetic information as classically defined by the central dogma of molecular biology. This course opens with discussions of the basic concepts and overview of classical examples of epigenetic phenomena in eukaryotes. The initial part of the course will set the framework to examine the various molecular mechanisms that have recently been uncovered in a number of eukaryotic models including yeast, Arabidopsis, maize, C. -

Human Biology - Genetics
Human Biology - Genetics The Program in Human Biology, Stanford University, (HumBio) Say Thanks to the Authors Click http://www.ck12.org/saythanks (No sign in required) www.ck12.org AUTHOR The Program in Human Biology, To access a customizable version of this book, as well as other Stanford University, (HumBio) interactive content, visit www.ck12.org CK-12 Foundation is a non-profit organization with a mission to reduce the cost of textbook materials for the K-12 market both in the U.S. and worldwide. Using an open-content, web-based collaborative model termed the FlexBook®, CK-12 intends to pioneer the generation and distribution of high-quality educational content that will serve both as core text as well as provide an adaptive environment for learning, powered through the FlexBook Platform®. Copyright © 2014 CK-12 Foundation, www.ck12.org The names “CK-12” and “CK12” and associated logos and the terms “FlexBook®” and “FlexBook Platform®” (collectively “CK-12 Marks”) are trademarks and service marks of CK-12 Foundation and are protected by federal, state, and international laws. Any form of reproduction of this book in any format or medium, in whole or in sections must include the referral attribution link http://www.ck12.org/saythanks (placed in a visible location) in addition to the following terms. Except as otherwise noted, all CK-12 Content (including CK-12 Curriculum Material) is made available to Users in accordance with the Creative Commons Attribution-Non-Commercial 3.0 Unported (CC BY-NC 3.0) License (http://creativecommons.org/ licenses/by-nc/3.0/), as amended and updated by Creative Com- mons from time to time (the “CC License”), which is incorporated herein by this reference. -

Population Genetics and Molecular Evolution - Session 1
Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics MSc in Bioinformatics Module 3: Genomics Lesson 6. Genome variation I. Theory and Data Antonio Barbadilla Group Genomics, Bioinformatics & Evolution Institut Biotecnologia I Biomedicina Departament de Genètica i Microbiologia UAB Bachelor’s Degree in Bioinformatics 1 Course 20122016-1317 Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics Population Genetics and Molecular Evolution Session 1. Introduction to Population Genetics Antonio Barbadilla Group Genomics, Bioinformatics & Evolution Institut Biotecnologia I Biomedicina Departament de Genètica i Microbiologia UAB Course 2018-19 Bachelor’s Degree in Bioinformatics 2 Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics Outline Darwinian evolution and Population Thinking Levels of genetic variation Population Genetics: the kinematics and dynamics of evolutionary changes in populations Types of genetic variation Gene and genotype frequencies Measures of nucleotide variation Surveys of genetic variation Population genetics databases The nature of genetic variation The golden age of population genetics Readings & exercices Bachelor’s Degree in Bioinformatics 3 Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics Why the rich diversity of life? Bachelor’s Degree in Bioinformatics Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics Bachelor’s Degree in Bioinformatics Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics 1859 Charles R. Darwin (1809-1882) Bachelor’s Degree in Bioinformatics Prof. Antonio Barbadilla Population Genetics and Molecular Evolution - Session 1. Introduction to Population Genetics Bachelor’s Degree in Bioinformatics Prof. -

Annexure-96. (Bsc Hons Biological Science)
दिल्ली दिश्िदिद्यालय UNIVERSITY OF DELHI Bachelor of Science (Hons) Biological Science (Effective from Academic Year 2019-20) Biological Science Sri Venkateswara College University of Delhi New Delhi-110021 Revised Syllabus as approved by Academic Council Date: No: Executive Council Date: No: Applicable for students registered with Regular Colleges List of Contents Page No. Preamble 1. Introduction to Programme …….(e.g. Biological Sciences Department) 1 2. Learning Outcome-based Curriculum Framework in Programme B.Sc. (Hons) Biological Science 1 2.1. Nature and Extent of the Programme in B.Sc. (Hons) Biological Science 2.2. Aims of Bachelor Degree Programme in B.Sc. (Hons) Biological Science 3. Graduate Attributes in B.Sc. (Hons) Biological Science 2 4. Qualification Descriptors for Graduates B.Sc. (Hons) Biological Science 3 5. Programme Learning Outcomes for in B.Sc. (Hons) Biological Science 4 6. Structure of in B.Sc. (Hons) Biological Science. 4 6.1. Credit Distribution for B.Sc. (Hons) Biological Science 6.2. Semester-wise Distribution of Courses. 7. Courses for Programme B.Sc. (Hons) Biological Science 9 PREAMBLE The undergraduate level education in basic science as well as in applied science has to be broad based. Well established disciplines like Physics, Chemistry, Mathematics, Botany, Zoology, Geology and many others have long developed interfaces with each other so much that areas like Chemical Physics, Biochemistry, Biophysics, Mathematical Biology or Geophysics have themselves emerged as major disciplines. Conceptualization in each of the above fundamental disciplines has made much of the information gathered meaningful. Mechanisms underlying many biological phenomena have been discovered. -

Silkworms, Science, and Nation: a Sericultural History of Genetics in Modern Japan
SILKWORMS, SCIENCE, AND NATION: A SERICULTURAL HISTORY OF GENETICS IN MODERN JAPAN A Dissertation Presented to the Faculty of the Graduate School of Cornell University in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Lisa Aiko Onaga January 2012 © 2012 Lisa Aiko Onaga SILKWORMS, SCIENCE, AND NATION: A SERICULTURAL HISTORY OF GENETICS IN MODERN JAPAN Lisa Aiko Onaga, Ph.D. Cornell University 2012 This dissertation describes how and why the source of raw silk, the domesticated silkworm (Bombyx mori), emerged as an organism that scientists in Japan researched intensively during the late nineteenth and early twentieth centuries. People invested in and exploited the lucrative silkworm in order to produce a delicate fiber, as well as to help impart universal claims and ideas about the governing patterns of inheritance at a time when uncertainties abounded about the principles of what we today call genetics. Silkworm inheritance studies such as those by scientists Toyama Kametarō (1867– 1918) and Tanaka Yoshimarō (1884–1972) contributed to ideas developing among geneticists internationally about the biological commonalities of different living organisms. Silkworm studies also interacted with the registration of silkworm varieties in and beyond East Asia at a time when the rising Imperial agenda intertwined with the silk industry. Different motivations drove silkworm science, apparent in the growth of Japanese understandings of natural order alongside the scientific pursuits of universality. Tōitsu, a “unification” movement around 1910, notably involved discussions about improving silk and decisions about the use of particular silkworms to generate export-bound Japanese silk. I show why the reasons for classifying silkworms within Japan had as much to do with the connection between textiles, power, and social order as it did with the turn toward experiment-based biological articulations of inheritance, which together interacted with ideas about Japanese nationhood.