Clades > 20 My
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brooklyn, Cloudland, Melsonby (Gaarraay)
BUSH BLITZ SPECIES DISCOVERY PROGRAM Brooklyn, Cloudland, Melsonby (Gaarraay) Nature Refuges Eubenangee Swamp, Hann Tableland, Melsonby (Gaarraay) National Parks Upper Bridge Creek Queensland 29 April–27 May · 26–27 July 2010 Australian Biological Resources Study What is Contents Bush Blitz? Bush Blitz is a four-year, What is Bush Blitz? 2 multi-million dollar Abbreviations 2 partnership between the Summary 3 Australian Government, Introduction 4 BHP Billiton and Earthwatch Reserves Overview 6 Australia to document plants Methods 11 and animals in selected properties across Australia’s Results 14 National Reserve System. Discussion 17 Appendix A: Species Lists 31 Fauna 32 This innovative partnership Vertebrates 32 harnesses the expertise of many Invertebrates 50 of Australia’s top scientists from Flora 62 museums, herbaria, universities, Appendix B: Threatened Species 107 and other institutions and Fauna 108 organisations across the country. Flora 111 Appendix C: Exotic and Pest Species 113 Fauna 114 Flora 115 Glossary 119 Abbreviations ANHAT Australian Natural Heritage Assessment Tool EPBC Act Environment Protection and Biodiversity Conservation Act 1999 (Commonwealth) NCA Nature Conservation Act 1992 (Queensland) NRS National Reserve System 2 Bush Blitz survey report Summary A Bush Blitz survey was conducted in the Cape Exotic vertebrate pests were not a focus York Peninsula, Einasleigh Uplands and Wet of this Bush Blitz, however the Cane Toad Tropics bioregions of Queensland during April, (Rhinella marina) was recorded in both Cloudland May and July 2010. Results include 1,186 species Nature Refuge and Hann Tableland National added to those known across the reserves. Of Park. Only one exotic invertebrate species was these, 36 are putative species new to science, recorded, the Spiked Awlsnail (Allopeas clavulinus) including 24 species of true bug, 9 species of in Cloudland Nature Refuge. -

Tasmanian Wilderness World Heritage Area
Appendix 4 1 World Heritage Values of the Tasmanian Wilderness 1.1 Note that the Department of the Environment's website states that: A draft Statement of Outstanding Universal Value which will take into account the new areas added in 2013 is expected to be considered by the World Heritage Committee in 2014. Outstanding Universal Value 1.2 The Tasmanian Wilderness is an extensive, wild, beautiful temperate land where cultural heritage of the Tasmanian Aboriginal people is preserved. 1.3 It is one of the three largest temperate wilderness areas remaining in the Southern Hemisphere. The region is home to some of the deepest and longest caves in Australia. It is renowned for its diversity of flora, and some of the longest lived trees and tallest flowering plants in the world grow in the area. The Tasmanian Wilderness is a stronghold for several animals that are either extinct or threatened on mainland Australia. 1.4 In the southwest Aboriginal people developed a unique cultural tradition based on a specialized stone and bone toolkit that enabled the hunting and processing of a single prey species (Bennett's wallaby) that provided nearly all of their dietary protein and fat. Extensive limestone cave systems contain rock art sites that have been dated to the end of the Pleistocene period. Southwest Tasmanian Aboriginal artistic expression during the last Ice Age is only known from the dark recesses of limestone caves. 1.5 The Tasmanian Wilderness was inscribed on the World Heritage List in 1982 and extended in 1989, 2010, 2012 and again in 2013. -

IV. on the Proteaceć of Jussieu. by Mr. Robert Brown, Lib. LS
IV. On the Proteacea of Jussieu. By -Mr. Robert Brown, Lib. L.S. Read Jan. 17, 1809. THELinnean system of botany, though confessedly artificial, has not only contributed more than all others to facilitate tlie knowledge of species, but, by constantly directing the attention to those essential parts of the flower on which it is founded, has made us acquainted with more of their important modific-a t’ ions than we probably should have known, had it not been generally adopted, and has thus laid a more solid foundation for the esta- blishment of a natural arrangement, the superior importance of which no one has been inore fully impressed with than Linnzus hiinself. There are still, however, certain circumstances respccting the stamina and pistilla, which appear to iiie to havc been much less attended to than they deserve, both by Linneus and succeeding botanists. What I chiefly allude to is the state of these organs before the expansion of the flower. Tlie utility of ascertaining the internal condition of the ovarium before fecundation will liardly be called in question, now that the immortal worlis of Gxrtner and Jussieu hare demonstrated the necessity of minutely studying the fruits of plants in attempting to arrange tlicin ac- cording to tlic sum of their affinities, as in many cases the true nature of tlie ripc fruit, cspecially witli respect to the placenta- tion of the seeds, can oiily be detcrniined by this mc;~ns. Its importance is indeed expressly inculcated by many l~ot:inists, Tf’llO, 16 Mr. BROWN,on the Proteacee of Jussieu. -

Street Tree Master Plan Report © Sunshine Coast Regional Council 2009-Current
Sunshine Coast Street Tree Master Plan 2018 Part A: Street Tree Master Plan Report © Sunshine Coast Regional Council 2009-current. Sunshine Coast Council™ is a registered trademark of Sunshine Coast Regional Council. www.sunshinecoast.qld.gov.au [email protected] T 07 5475 7272 F 07 5475 7277 Locked Bag 72 Sunshine Coast Mail Centre Qld 4560 Acknowledgements Council wishes to thank all contributors and stakeholders involved in the development of this document. Disclaimer Information contained in this document is based on available information at the time of writing. All figures and diagrams are indicative only and should be referred to as such. While the Sunshine Coast Regional Council has exercised reasonable care in preparing this document it does not warrant or represent that it is accurate or complete. Council or its officers accept no responsibility for any loss occasioned to any person acting or refraining from acting in reliance upon any material contained in this document. Foreword Here on our healthy, smart, creative Sunshine Coast we are blessed with a wonderful environment. It is central to our way of life and a major reason why our 320,000 residents choose to live here – and why we are joined by millions of visitors each year. Although our region is experiencing significant population growth, we are dedicated to not only keeping but enhancing the outstanding characteristics that make this such a special place in the world. Our trees are the lungs of the Sunshine Coast and I am delighted that council has endorsed this master plan to increase the number of street trees across our region to balance our built environment. -
Abelmoschus Moschatus Subsp
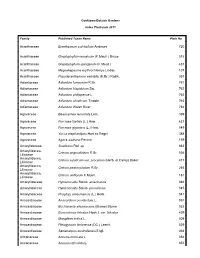
Cooktown Botanic Gardens Index Plantarum 2011 Family Published Taxon Name Plate No Acanthaceae Eranthemum pulchellum Andrews 720 Acanthaceae Graptophyllum excelsum (F.Meull.) Druce 515 Acanthaceae Graptophyllum spinigerum (F.Meull.) 437 Acanthaceae Megaskepasma erythrochlamys Lindau 107 Acanthaceae Pseuderanthemum variabile (R.Br.) Radlk. 357 Adiantaceae Adiantum formosum R.Br. 761 Adiantaceae Adiantum hispidulum Sw. 762 Adiantaceae Adiantum philippense L. 765 Adiantaceae Adiantum silvaticum Tindale 763 Adiantaceae Adiantum Walsh River 764 Agavaceae Beaucarnea recurvata Lem. 399 Agavaceae Furcraea foetida (L.) Haw. 637 Agavaceae Furcraea gigantea (L.) Haw. 049 Agavaceae Yucca elephantipes Hort.ex Regel 388 Agavaceae Agave sisalana Perrine. 159 Amarylidaceae Scadoxus Raf. sp 663 Amaryllidacea, Crinum angustifolium R.Br. 536 Liliaceae Amaryllidacea, Crinum asiaticum var. procerum (Herb. et Carey) Baker 417 Liliaceae Amaryllidacea, Crinum pedunculatum R.Br. 265 Liliaceae Amaryllidacea, Crinum uniflorum F.Muell. 161 Liliaceae Amaryllidaceae Hymenocallis Salisb. americanus 046 Amaryllidaceae Hymenocallis Salisb. peruvianna 045 Amaryllidaceae Proiphys amboinensis (L.) Herb. 041 Anacardiaceae Anacardium occidentale L. 051 Anacardiaceae Buchanania arborescens (Blume) Blume. 022 Anacardiaceae Euroschinus falcatus Hook.f. var. falcatus 429 Anacardiaceae Mangifera indica L. 009 Anacardiaceae Pleiogynium timorense (DC.) Leenh. 029 Anacardiaceae Semecarpus australiensis Engl. 368 Annonaceae Annona muricata L. 054 Annonaceae Annona reticulata L. 053 Annonaceae Annona squamosa 602 Annonaceae Cananga odorata (Lam.) Hook.f.&Thomson 406 Annonaceae Melodorum leichhardtii (F.Muell.) Diels. 360 Annonaceae Rollinia deliciosa Saff. 098 Apiaceae Centella asiatica (L.) Urb. 570 Apocynaceae Adenium obesum (Forssk.) Roem. & Schult. 489 Apocynaceae Allamanda cathartica L. 047 Apocynaceae Allamanda violacea Gardn. & Field. 048 Apocynaceae Alstonia actinophylla (A.Cunn.) K.Schum. 026 Apocynaceae Alstonia scholaris (L.) R.Br. 012 Apocynaceae Alyxia ruscifolia R.Br. -

Finschia-"A Genus of "Nut" Trees of the Southwest Pacific
Finschia-" A Genus of "Nut" Trees of the Southwest Pacific c. T. WHITE1 INTRODUCTION A PLANT FAMILY with a most interesting and F. Muell., Carnarvonia F. Muell., D arlin"gia F; intriguing distribution is Proteaceae, which finds Muell., Hollandaea F. Muell. (two spp.) , Mus its greatest development in Australia (650 " gravea F. Muell., and Placospermum White & species) on the one hand and South Africa (300 Francis. A surprising feature is the absence, species) on the other, though the two countries with the exception of one species in New Zea have no genera in common. Practically all the land, of the family "from Polynesia. South African species and the vast majority of There is in the islands of the southwest Paci "Australian ones are markedly xerophytic. The fic-Caroline Islands, New Guinea, Solomon largest genus, Greoillea R. Br., consists mainly Islands, and the New Hebrides-a group of trees of xerophytic shrubs or small trees but a few with the floral characters of Greuillea R. Br. are large trees found in the rain forests of and the fruit of Helicia Lour. These, I consider, tropical and subtropical eastern Australia, New all belong 'to Finschia Warb. This genus was Guinea, and New Caledonia. In the southwest founded by Warburg (1891: 297 ) on"a tree Pacific area the family finds its greatest develop from northeastern New Guinea. His original ment in northeastern Australia, where trees be description would cover Grevillea R. Br. exactly longing to it provide the great bulk of cabinet though he does not mention this genus and on timbers known in the trade as "Silky Oaks." the following page the distinctions he gives for There is close affinity between the Proteaceae of separating his proposed new genus from H elicia eastern Australia and of western South America are exactly those which distinguish Greuillea as illustrated by the genera Embothrium Forst. -

Pathogens Associated with Diseases. of Protea, Leucospermum and Leucadendron Spp
PATHOGENS ASSOCIATED WITH DISEASES. OF PROTEA, LEUCOSPERMUM AND LEUCADENDRON SPP. Lizeth Swart Thesis presented in partial fulfillment of the requirements for the degree of Master of Science in Agriculture at the University of Stellenbosch Supervisor: Prof. P. W. Crous Decem ber 1999 Stellenbosch University https://scholar.sun.ac.za DECLARATION 1, the undersigned, hereby declare that the work contained in this thesis is my own original work and has not previously in its entirety or in part been submitted at any university for a degree. SIGNATURE: DATE: Stellenbosch University https://scholar.sun.ac.za PATHOGENS ASSOCIATED WITH DISEASES OF PROTEA, LEUCOSPERMUM ANDLEUCADENDRONSPP. SUMMARY The manuscript consists of six chapters that represent research on different diseases and records of new diseases of the Proteaceae world-wide. The fungal descriptions presented in this thesis are not effectively published, and will thus be formally published elsewhere in scientific journals. Chapter one is a review that gives a detailed description of the major fungal pathogens of the genera Protea, Leucospermum and Leucadendron, as reported up to 1996. The pathogens are grouped according to the diseases they cause on roots, leaves, stems and flowers, as well as the canker causing fungi. In chapter two, several new fungi occurring on leaves of Pro tea, Leucospermum, Telopea and Brabejum collected from South Africa, Australia or New Zealand are described. The following fungi are described: Cladophialophora proteae, Coniolhyrium nitidae, Coniothyrium proteae, Coniolhyrium leucospermi,Harknessia leucospermi, Septoria prolearum and Mycosphaerella telopeae spp. nov. Furthermore, two Phylloslicla spp., telopeae and owaniana are also redecribed. The taxonomy of the Eisinoe spp. -

Table of Contents Below) with Family Name Provided
1 Australian Plants Society Plant Table Profiles – Sutherland Group (updated August 2021) Below is a progressive list of all cultivated plants from members’ gardens and Joseph Banks Native Plants Reserve that have made an appearance on the Plant Table at Sutherland Group meetings. Links to websites are provided for the plants so that further research can be done. Plants are grouped in the categories of: Trees and large shrubs (woody plants generally taller than 4 m) Medium to small shrubs (woody plants from 0.1 to 4 m) Ground covers or ground-dwelling (Grasses, orchids, herbaceous and soft-wooded plants, ferns etc), as well as epiphytes (eg: Platycerium) Vines and scramblers Plants are in alphabetical order by botanic names within plants categories (see table of contents below) with family name provided. Common names are included where there is a known common name for the plant: Table of Contents Trees and Large shrubs........................................................................................................................... 2 Medium to small shrubs ...................................................................................................................... 23 Groundcovers and other ground‐dwelling plants as well as epiphytes. ............................................ 64 Vines and Scramblers ........................................................................................................................... 86 Sutherland Group http://sutherland.austplants.com.au 2 Trees and Large shrubs Acacia decurrens -

Plant Life of Western Australia
INTRODUCTION The characteristic features of the vegetation of Australia I. General Physiography At present the animals and plants of Australia are isolated from the rest of the world, except by way of the Torres Straits to New Guinea and southeast Asia. Even here adverse climatic conditions restrict or make it impossible for migration. Over a long period this isolation has meant that even what was common to the floras of the southern Asiatic Archipelago and Australia has become restricted to small areas. This resulted in an ever increasing divergence. As a consequence, Australia is a true island continent, with its own peculiar flora and fauna. As in southern Africa, Australia is largely an extensive plateau, although at a lower elevation. As in Africa too, the plateau increases gradually in height towards the east, culminating in a high ridge from which the land then drops steeply to a narrow coastal plain crossed by short rivers. On the west coast the plateau is only 00-00 m in height but there is usually an abrupt descent to the narrow coastal region. The plateau drops towards the center, and the major rivers flow into this depression. Fed from the high eastern margin of the plateau, these rivers run through low rainfall areas to the sea. While the tropical northern region is characterized by a wet summer and dry win- ter, the actual amount of rain is determined by additional factors. On the mountainous east coast the rainfall is high, while it diminishes with surprising rapidity towards the interior. Thus in New South Wales, the yearly rainfall at the edge of the plateau and the adjacent coast often reaches over 100 cm. -

Evolutionary History of Floral Key Innovations in Angiosperms Elisabeth Reyes
Evolutionary history of floral key innovations in angiosperms Elisabeth Reyes To cite this version: Elisabeth Reyes. Evolutionary history of floral key innovations in angiosperms. Botanics. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS489. tel-01443353 HAL Id: tel-01443353 https://tel.archives-ouvertes.fr/tel-01443353 Submitted on 23 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS489 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de Doctorat : Biologie Par Mme Elisabeth Reyes Evolutionary history of floral key innovations in angiosperms Thèse présentée et soutenue à Orsay, le 13 décembre 2016 : Composition du Jury : M. Ronse de Craene, Louis Directeur de recherche aux Jardins Rapporteur Botaniques Royaux d’Édimbourg M. Forest, Félix Directeur de recherche aux Jardins Rapporteur Botaniques Royaux de Kew Mme. Damerval, Catherine Directrice de recherche au Moulon Président du jury M. Lowry, Porter Curateur en chef aux Jardins Examinateur Botaniques du Missouri M. Haevermans, Thomas Maître de conférences au MNHN Examinateur Mme. Nadot, Sophie Professeur à l’Université Paris-Sud Directeur de thèse M. -

Attachment E - Desktop Searches
Attachment E - Desktop searches EPBC Act Protected Matters Report This report provides general guidance on matters of national environmental significance and other matters protected by the EPBC Act in the area you have selected. Information on the coverage of this report and qualifications on data supporting this report are contained in the caveat at the end of the report. Information is available about Environment Assessments and the EPBC Act including significance guidelines, forms and application process details. Report created: 11/06/20 13:02:49 Summary Details Matters of NES Other Matters Protected by the EPBC Act Extra Information Caveat Acknowledgements This map may contain data which are ©Commonwealth of Australia (Geoscience Australia), ©PSMA 2010 Coordinates Buffer: 20.0Km Summary Matters of National Environmental Significance This part of the report summarises the matters of national environmental significance that may occur in, or may relate to, the area you nominated. Further information is available in the detail part of the report, which can be accessed by scrolling or following the links below. If you are proposing to undertake an activity that may have a significant impact on one or more matters of national environmental significance then you should consider the Administrative Guidelines on Significance. World Heritage Properties: None National Heritage Places: None Wetlands of International Importance: None Great Barrier Reef Marine Park: None Commonwealth Marine Area: None Listed Threatened Ecological Communities: 4 Listed Threatened Species: 26 Listed Migratory Species: 16 Other Matters Protected by the EPBC Act This part of the report summarises other matters protected under the Act that may relate to the area you nominated. -

TELOPEA Publication Date: 27 September 1991 Til
Volume 4(3): 497–507 TELOPEA Publication Date: 27 September 1991 Til. Ro)'al BOTANIC GARDENS dx.doi.org/10.7751/telopea19914946 Journal of Plant Systematics 6 DOPII(liPi Tmst plantnet.rbgsyd.nsw.gov.au/Telopea • escholarship.usyd.edu.au/journals/index.php/TEL· ISSN 0312-9764 (Print) • ISSN 2200-4025 (Online) 497 Alloxylon (Proteaceae), a new genus from New Guinea and eastern Australia Peter H. Weston and Michael D. Crisp Abstract Weston, Peter H.I, and Crisp, Michael D.2 (1 National Herbarium of New South Wales, Royal Botanic Gardens, Sydney NSW Australia 2000; 2 Australian National Botanic Gardens, GPO Box 1777, Canberra ACT Australia 2601; present address: Division of Botany and Zoology, Australian National University, GPO Box 4, Canberra ACT 2601) 1991. Alloxylon (Proteaceae), a new genus from New Guinea and eastern Australia. Telopea 4(3): 497-507. Oreocallis sens. lat. consists of two distinct clades, one in South America, the other in Australasia, that together are likely to be paraphylet ic. Newly sampled characters strongly support the monophyly of the Australasian group. We describe the new genus Alloxylon to accommodate the Australasian species of Oreocallis sens. lat. and revise its species. Alloxylon fIammeum is described as new and new combinations are made for A. brachycarpum, A. wickhamii and A. pinnatum. Introduction Cladistic analyses of the subtribe Embothriinae of the family Proteaceae (Weston & Crisp 1987, in prep.) show the genus Oreocallis R. Br. sens. lat. to comprise two branches of an unresolved trichotomy. The third branch is the genus Telopea, a well corroborated clade of five species. The sister group to this trichotomy is Embothrium, the only other genus in the Embothriinae.