European Patent Office of Opposition to That Patent, in Accordance with the Implementing Regulations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Determination of Iodate in Iodised Salt by Redox Titration
College of Science Determination of Iodate in Iodised Salt by Redox Titration Safety • 0.6 M potassium iodide solution (10 g solid KI made up to 100 mL with distilled water) • 0.5% starch indicator solution Lab coats, safety glasses and enclosed footwear must (see below for preparation) be worn at all times in the laboratory. • 250 mL volumetric flask Introduction • 50 mL pipette (or 20 and 10 mL pipettes) • 250 mL conical flasks New Zealand soil is low in iodine and hence New Zealand food is low in iodine. Until iodised salt was • 10 mL measuring cylinder commonly used (starting in 1924), a large proportion • burette and stand of school children were reported as being affected • distilled water by iodine deficiency – as high as 60% in Canterbury schools, and averaging 20 − 40% overall. In the worst cases this deficiency can lead to disorders such as Method goitre, and impaired physical and mental development. 1. Preparation of 0.002 mol L−1 sodium thiosulfate In earlier times salt was “iodised” by the addition of solution: Accurately weigh about 2.5 g of solid potassium iodide; however, nowadays iodine is more sodium thiosulfate (NaS2O3•5H2O) and dissolve in commonly added in the form of potassium iodate 100 mL of distilled water in a volumetric flask. (This gives a 0.1 mol L−1 solution). Then use a pipette to (KIO3). The Australia New Zealand Food Standards Code specifies that iodised salt must contain: “equivalent to transfer 10 mL of this solution to a 500 mL volumetric no less than 25 mg/kg of iodine; and no more than 65 flask and dilute by adding distilled water up to the mg/kg of iodine”. -

WHO Model List of Essential Medicines
WHO Model List of Essential Medicines 15th list, March 2007 Status of this document This is a reprint of the text on the WHO Medicines web site http://www.who.int/medicines/publications/essentialmedicines/en/index.html 15th edition Essential Medicines WHO Model List (revised March 2007) Explanatory Notes The core list presents a list of minimum medicine needs for a basic health care system, listing the most efficacious, safe and cost‐effective medicines for priority conditions. Priority conditions are selected on the basis of current and estimated future public health relevance, and potential for safe and cost‐effective treatment. The complementary list presents essential medicines for priority diseases, for which specialized diagnostic or monitoring facilities, and/or specialist medical care, and/or specialist training are needed. In case of doubt medicines may also be listed as complementary on the basis of consistent higher costs or less attractive cost‐effectiveness in a variety of settings. The square box symbol () is primarily intended to indicate similar clinical performance within a pharmacological class. The listed medicine should be the example of the class for which there is the best evidence for effectiveness and safety. In some cases, this may be the first medicine that is licensed for marketing; in other instances, subsequently licensed compounds may be safer or more effective. Where there is no difference in terms of efficacy and safety data, the listed medicine should be the one that is generally available at the lowest price, based on international drug price information sources. Therapeutic equivalence is only indicated on the basis of reviews of efficacy and safety and when consistent with WHO clinical guidelines. -

United States Patent Office Patented Apr
3,505,222 United States Patent Office Patented Apr. 7, 1970 1. 2 3,505,222 product of a mercaptain with sulfur trioxide. Their metal LUBRICANT COMPOSITIONS salts are represented by the formula: Leonard M. Niebylski, Birmingham, Mich, assignor to O Ethyl Corporation, New York, N.Y., a corporation of Virginia (R-S-S-0--M No Drawing. Filed Mar. 29, 1967, Ser. No. 626,701 5 s (I) Int. C. C10m 5/14, 3/18, 7/36 wherein R is a hydrocarbon radical containing from 1 U.S. C. 252-17 2 Claims to about 30 carbon atoms, M is a metal, and n is the valence of metal M. For example, when M is the monova 0. lent sodium ion, n is 1. ABSTRACT OF THE DISCLOSURE The radical R can be an alkyl, cycloalkyl, aralkyl, The extreme pressure wear properties of base lubri alkaryl, or aryl radical. The radicals may contain other cants including water, hydrocarbons, polyesters, silicones, nonhydrocarbon substituents such as chloro, bromo, iodo, polyethers and halocarbons is enhanced by the addition fluoro, nitro, hydroxyl, nitrile, isocyanate, carboxyl, car of a synergistic mixture of a thiosulfate compound and 15 bonyl, and the like. a lead compound. The useful metals are all those capable of forming Bunte salts. Preferred metals are those previously listed as suitable for forming metal thiosulfates. Of these, the Background more preferred metals are sodium and lead, and lead is 20 the most preferred metal in the Bunte salts. This invention relates to improved lubricant composi Examples of useful Bunte salts include: tions. -

Sodium Nitrite and Sodium Thiosulfate
PATIENT & CAREGIVER EDUCATION Sodium Nitrite and Sodium Thiosulfate This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Nithiodote Warning This drug may cause low blood pressure and a red blood cell problem called methemoglobinemia. These may be life-threatening. This drug is only for use when cyanide poisoning is life-threatening. This drug must be used with care if it is not known if cyanide poisoning has happened. Talk with the doctor. Tell the doctor if your child has inhaled a lot of smoke or if your child has any of these health problems: Anemia, heart problems, lack of a certain enzyme called congenital methemoglobin reductase deficiency, or lung problems. What is this drug used for? It is used to treat cyanide poisoning. What do I need to tell the doctor BEFORE my child takes this drug? If your child is allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell the doctor about the allergy and what signs your child had. Sodium Nitrite and Sodium Thiosulfate 1/6 If your child is breast-feeding a baby: Be sure your child does not breast-feed a baby while taking this drug. This drug may interact with other drugs or health problems. Tell the doctor and pharmacist about all of your child’s drugs (prescription or OTC, natural products, vitamins) and health problems. You must check to make sure that it is safe to give this drug with all of your child’s other drugs and health problems. -

Assessing the in Situ Efficacy of Tea Tree Oil As a Topical Antiseptic
Novasel Australia Pty Ltd Assessing the in situ efficacy of tea tree oil as a topical antiseptic A report for the Rural Industries Research and Development Corporation by S. Messager, K.A. Hammer & T.V. Riley August 2005 RIRDC Publication No 05/113 RIRDC Project No UWA-72A © 2005 Rural Industries Research and Development Corporation. All rights reserved. ISBN 1 74151 176 3 ISSN 1440-6845 Assessing the in situ efficacy of tea tree oil as a topical antiseptic Publication No. 05/113 Project No. UWA-72A The information contained in this publication is intended for general use to assist public knowledge and discussion and to help improve the development of sustainable industries. The information should not be relied upon for the purpose of a particular matter. Specialist and/or appropriate legal advice should be obtained before any action or decision is taken on the basis of any material in this document. The Commonwealth of Australia, Rural Industries Research and Development Corporation, the authors or contributors do not assume liability of any kind whatsoever resulting from any person's use or reliance upon the content of this document. This publication is copyright. However, RIRDC encourages wide dissemination of its research, providing the Corporation is clearly acknowledged. For any other enquiries concerning reproduction, contact the Publications Manager on phone 02 6272 3186. Researcher Contact Details Prof. T. V. Riley University of Western Australia School of Biomedical and Chemical Sciences Microbiology (M502) 35 Stirling Hwy CRAWLEY WA 6009 Phone: (08) 9346 3690 Fax: (08) 9346 2912 Email: [email protected] In submitting this report, the researcher has agreed to RIRDC publishing this material in its edited form. -

Chemical %Concentration CAS
Sebozole Shampoo Page 1of 5 Sebozole Shampoo SAFETY DATA SHEET (1) IDENTIFICATION Name: Sebozole Shampoo Use: veterinary skin care Supplier: Vetoquinol USA (Tomlyn Products) 4250 N. Sylvania Ave Fort Worth, TX 76137 Tel: ( 817)529-7500 Fax: (817)529-7506 (2) HAZARD(S) IDENTIFICATION Hazard Classification: Non Hazardous as defined by 29CFR Part 1910.1200 (3) COMPOSITION/INFORMATION ON INGREDIENTS Common Name: Sebozole Shampoo Composition: A blend of proprietary ingredients which make a veterinary dermatologic care product. Chemical %Concentration CAS Sodium Thiosulfate 3.13 10102-17-7 Miconazole Nitrate 2.0 22916-47-8 Salicylic Acid 2.0 69-72-7 Chloroxylenol 1.0 88-04-0 Sodium Olefin Sulfonate 25 6843-57-6 PEG 150 Pentaerythrityl Tetratstearate 5 130249-48-8 Lauramide DEA 5 92680-75-6 Propylene Glycol 5 57-55-6 Ultrez 21 1 9003-39-8 Sodium Hydroxide 1 1310-73-2 Fragrance 0.5 Kathon CG 0.1 6118-96-6 FD&C yellow #5 0.0013 84842-94-4 FD&C Blue #1 0.0013 3844-45-9 Purified Water 49 7732-18-5 Sebozole Shampoo Page 2 of 5 (4) First Aid Measures Ingestion • In case of ingestion call a physician and poison control. EYE • If product enters eye, rinse thouroughly with cool fresh water for 10 to 15 minutes. If irritation persists, seek medical attention. • a burning sensation, excessive tears, sensitivity to light, swelling and redness of the conjunctiva and increasedblinking. • Limited evidence or practical experience suggests, that the material may cause eye irritation in a substantial number of individuals.Prolonged eye contact may cause inflammation characterized by a redness of the conjunctiva (similar to windburn SKIN • none INHALED • None (5) FIRE-FIGHTING MEASURES Suitable Extinguisher: FOAM, DRY POWDER, WATER, CO2 Special Precautions: None Special Protective Equipment: None (6) ACCIDENTAL RELEASE PROCEDURES Personal Precautions: None. -
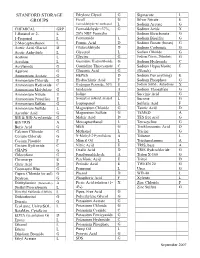
Stanford Storage Groups
STANFORD STORAGE Ethylene Glycol G Sigmacote L GROUPS Ficoll G Silver Nitrate E Formaldehyde w/ methanol L Sodium Acetate G CHEMICAL GRP Formaldehyde <37%, G Sodium Azide X 1-Butanol or 2- L 20% NBF Formalin Sodium Bicarbonate G 1-Propanol L Formamide L Sodium Bisulfite G 2-Mercaptoethanol L Formic Acid D Sodium Borate (borax) G Acetic Acid, Glacial D Glutaraldehyde D Sodium Carbonate G Acetic Anhydride L Glycerol L Sodium Chloride G Acetone L Glycine G Sodium Citrate, Dihydrate G Acrolein L Guanidine Hydrochloride G Sodium Hydroxide C Acrylamide G Guanidine Thiocyanate C Sodium Hypochlorite E Agarose G Gluconic Acid G (Bleach) Ammonium Acetate G HEPES D Sodium Per(anything) E Ammonium Chloride G Hydrochloric Acid F Sodium Phosphate G Ammonium Hydroxide C Hydrogen Peroxide, 30% E Sodium Sulfide, Anhydrous X Ammonium Molybdate G Imidazole A Sodium Thiosulfate G Ammonium Nitrate E Iodine E Succinic Acid G Ammonium Persulfate E Isoamyl or isobutyl alcohol L Sucrose G Ammonium Sulfate G Isopropanol L Sulfuric Acid F Ammonium Sulfide L Magnesium Chloride G Tannic Acid D Ascorbic Acid G Magnesium Sulfate G TEMED A BIS & BIS-Acrylamide G Maleic Acid D TES free acid G BIS TRIS A Mercaptoethanol L Tetracycline G Boric Acid G MES G Trichloroacetic Acid D Calcium Chloride G Methanol L Tricine G Cesium Chloride G N-Methyl-2-Pyrrolidone A Toluene L Cesium Fluoride F Mineral Oil L Triethanolamine A Cesium Hydroxide C Nitric Acid E TRIS, base A CHAPS G Oxalic Acid D TRIS Hydrochloride G Chloroform G Paraformaldehyde A Triton X-100 G Chromerge E Perchloric -

PHARMACEUTICAL STARTING MATERIALS List of Products
PHARMACEUTICAL STARTING MATERIALS List of Products Acetylsalicylic Acid, Dextrose Anhydrous Ketamine Hydrochloride Povidone crystalline Acyclovir Diazepam Ketoconazole Pravastain Albendazole Diclofenac Potasium Ketoprofen Praziquantel Allopurinol Diclofenac Sodium Labetalol Prednisolone Aluminium Hydroxide Difloxacin Lamivudine Prednisolone Hydrochloride Acetate Amikacin Sulfate Dihydroartemisinin Levamisole Procaine Benzylpenicillin, sterile powder Aminophylline Dihydostreptomycin Levofloxacin Sterile Procaine Sulfate Powder Hydrochloride Amodiaquine Diloxanide Furoate Levonorgestrel Progesterone Hydrochloride Amoxicillin Trihydrate, Diphenhydramine Levothyroxine Sodium Promethazine non-sterile powder, Hydrochloride Hydrochloride compact Ampicillin Sodium, Doxazosin Mesylate Lidocaine Hydrochloride Propranolol sterile crystalline Hydrochloride Ampicillin Trihydrate, Doxycycline Hyclate Lincomycin Pyrantel Pamoate non-sterile powder Hydrochloride Ampicillin Trihydrate, Doxycycline Lisinopril Pyrazinamide compact powder Monohydrate Ascorbic Acid Econazole Nitrate L-Methionine Pyridoxine Hydrochloride, powder Atropine Sulfate Eflorinthine Loratadine Pyrimethamine Hydrochloride Azithromycin Enalapril Maleate Lovastatin Quinine Hydrochloride Benzanthine Enrofloxacin Magnesium Trisilicate Ramipril Benzylpenicillin, sterile Hydrochloride powder Benzylpenicillin Epinephrine Magnesium Sulfate, Ranitidine Potassium, sterile dried powder Hydrochloride powder Benzylpenicillin Ergocalciferol Mannitol Retinol Acetate Sodium, sterile powder Betamethasone -

WHO Model List (Revised April 2003) Explanatory Notes
13th edition (April 2003) Essential Medicines WHO Model List (revised April 2003) Explanatory Notes The core list presents a list of minimum medicine needs for a basic health care system, listing the most efficacious, safe and cost-effective medicines for priority conditions. Priority conditions are selected on the basis of current and estimated future public health relevance, and potential for safe and cost-effective treatment. The complementary list presents essential medicines for priority diseases, for which specialized diagnostic or monitoring facilities, and/or specialist medical care, and/or specialist training are needed. In case of doubt medicines may also be listed as complementary on the basis of consistent higher costs or less attractive cost-effectiveness in a variety of settings. When the strength of a drug is specified in terms of a selected salt or ester, this is mentioned in brackets; when it refers to the active moiety, the name of the salt or ester in brackets is preceded by the word "as". The square box symbol (? ) is primarily intended to indicate similar clinical performance within a pharmacological class. The listed medicine should be the example of the class for which there is the best evidence for effectiveness and safety. In some cases, this may be the first medicine that is licensed for marketing; in other instances, subsequently licensed compounds may be safer or more effective. Where there is no difference in terms of efficacy and safety data, the listed medicine should be the one that is generally available at the lowest price, based on international drug price information sources. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Senna Alata (Akapulko) Extract Versus Topical Antifungals for Treatment of Superficial Fungal Skin Infections: a Systematic Review and Meta-Analysis
ORIGINAL ARTICLE Senna alata (akapulko) Extract versus Topical Antifungals for Treatment of Superficial Fungal Skin Infections: a Systematic Review and Meta-analysis Erin Jane L. Tababa,1 Rowena Natividad S. Flores-Genuino2 and Charissa Mia D. Salud-Gnilo1 1Section of Dermatology, Department of Medicine, College of Medicine and Philippine General Hospital, University of the Philippines Manila 2Department of Anatomy, College of Medicine, University of the Philippines Manila ABSTRACT Objective. The study aimed to assess the efficacy and safety of Senna alata (akapulko) plant extracts compared with topical antifungals in the treatment of superficial fungal skin infections. Methods. A systematic review and meta-analysis of randomized controlled trials that studied patients with diagnosed cutaneous tinea or dermatophytosis (excluding hair and nail), tinea versicolor, or cutaneous candidiasis, via microscopy or culture, and compared the efficacy and safety of S. alata (akapulko) extract versus topical antifungals. Two authors independently screened titles and abstracts of merged search results from electronic databases (The Cochrane Skin Group Specialized Register, CENTRAL, MEDLINE, EMBASE (January 1990 to December 2011), Health Research and Development Information Network (HERDIN), and reference lists of articles), assessed eligibility, assessed the risk of bias using the domains in the Cochrane Risk Bias tool and collected data using a pretested Data extraction form (DEF). Meta-analyses were performed when feasible. Results. We included seven RCTs in the review. There is low certainty of evidence that S. alata 50% lotion is as efficacious as sodium thiosulfate 25% lotion (RR 0.91, 95% CI, 0.79 to 1.04; 4 RCTs, n=216; p=0.15;2 I =52%) and high quality evidence that S.