Trans-Scale-Scope to Find Rare Cellular Activity in Sub-Million Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

What Is Osaka University Doing in ASEAN Countries?
Osaka University ASEAN Center for Academic Initiatives Contact address: Room C, 10th Floor, Serm-Mit Tower, Sukhumvit 21 (Soi Asok), Klongtoey-nua, Wattana, Bangkok 10110, Thailand http://www.bangkok.overseas.osaka-u.ac.jp/ Osaka University was founded in 1931 as the 6th imperial university of Japan through strong demand from the business and government sectors of Osaka, as well as the people of Osaka City and Prefecture. Now Osaka University holds 11 schools,16 graduate schools and 25 research centers and institutes with 15,250 undergraduate and 8,054 graduate students including 2,480 international students and 3,541 full-time academic and 3,113 full- time non-academic staff. It also has 4 libraries and 2 university hospitals; 3 campuses in Suita, Toyonaka, and Minoh. What is Osaka University doing in ASEAN countries? ASEAN Center for Academic Initiatives is one of the four overseas centers of Osaka University. The mission is to bridge Osaka U with ASEAN universities and academics, and to encourage “study in Osaka University” to ASEAN students. International Center for Biotechnology operates the Collaborative Research Center for Bioscience and Biotechnology in Mahidol University to promote joint researches with Thai universities such as CU, MU, KU and KMUTT, and offers the international educational programs under the support of Japanese Government, UNESCO and Thai universities. Research Institute for Microbial Diseases operates Thailand-Japan Research Collaboration Center on Emerging and Re-emerging Infections (RCC-ERI) under the Japanese national project. RCC-ERI carries out the qualified researches on infectious bacteria and viruses specific in tropical region. It has also research laboratories in Mahidol University. -

Clinical Trial of Step-Up Exercise Therapy Using DVD for Patients with Hip Osteoarthritis
Abstracts / Osteoarthritis and Cartilage 20 (2012) S54–S296 S265 Symmetry Ratios (Operated / Non-Operated Limb) Symmetry Group (Pre-TKA) Symmetry Group (Post-Rehabilitation) Strengthening Group (Post-Rehabilitation) Knee Flexion Excursion Symmetry Ratio 0.5 +/- 0.3 1.1 +/- 1.0 0.7 +/- 0.4 Peak Knee Flexion Moment Symmetry Ratio 0.7 +/- 0.6 1.2 +/- 0.8 0.8 +/- 0.5 Peak Knee Flexion During Stance Symmetry Ratio 0.8 +/- 0.8 1.0 +/- 0.3 0.8 +/- 0.2 523 comments were also related with encouragement of correct under- CLINICAL TRIAL OF STEP-UP EXERCISE THERAPY USING DVD FOR standing of exercise using DVD. PATIENTS WITH HIP OSTEOARTHRITIS Y. Uesugi 1, J. Koyanagi 2, T. Nishii 2, S. Hayashi 3, T. Fujishiro 3, K. Takagi 4,R. Yamaguchi 5, T. Nishiyama 3. 1 Kobe Univ. Graduate Sch. of Hlth.Sci., Kobe, Japan; 2 Osaka Univ. Graduate Sch. of Med., Suita, Japan; 3 Kobe Univ. Sch. of Med., Kobe, Japan; 4 Dept. of Rehabilitation, Osaka Univ. Hosp., Suita, Japan; 5 Div. of Rehabilitation, Kobe Univ. Hosp., Kobe, Japan Purpose: Efficacy of exercise treatment for osteoarthritis (OA) has been reported controversially. We developed a new exercise program designed as several step-up stages in accordance with patient disease activity, using a digital video disc (DVD). In this study, we analyzed the efficacy of this step-up exercise therapy program using DVD for the hip OA patients. Methods: We prospectively recruited 45 patients (male1, female44) with symptomatic hip OA were included. All participants provided informed consent to the study which was approved by the Institutional Review Board. -

Storm Warning (Bofu-Keiho / 暴 風警報) Or an Emergency Warning (Tokubetsu-Keiho / 特別警報)
Class Cancellation due to Weather Warnings: Storm Warning (Bofu-keiho / 暴 風警報) or an Emergency Warning (Tokubetsu-keiho / 特別警報) At the moment, a typhoon is approaching Japan. Classes will be cancelled if any of the above warnings are issued. You can confirm the details of when class cancellation may occur according to areas and municipalities where warnings have been issued, and when the warning has been lifted on the following homepage or the table below. Kwansei Gakuin University Website Undergraduate: http://www.kwansei.ac.jp/a_affairs/a_affairs_000850.html Graduate : http://www.kwansei.ac.jp/a_affairs/a_affairs_002656.html Nishinomiya-Uegahara and Kobe-Sanda Warning/Strike Lifted Nishinomiya-Seiwa Campus Campus By 6:00 am All classes held as usual 1st period class cancelled By 8:00 am Both 2nd-5th period class held as usual Undergraduate 1st & 2nd period classes cancelled By 10:30 am All classes and Graduate 3rd - 5th period classes held as usual cancelled School 1st - 3rd period classes cancelled By 12:00 pm 4th - 5th period classes held as usual Any time after 12:00 pm All classes cancelled 1st - 5th period classes cancelled Graduate By 3:00 pm 6th – 7th period classes held as usual School only Any time after 3:00 pm All classes cancelled Areas Municipalities Hanshin Kobe, Amagasaki, Nishinomiya, Ashiya, Itami, Takarazuka, Kawanishi, Sanda, Inagawa Hokuban Tanba Nishiwaki, Sasayama, Tanba, Taka-cho Harima Nantobu Akashi, Kakogawa, Miki, Takasago, Ono, Kasai, Kato, Inami-cho, Harima-cho Osaka Osaka city Kita Osaka Toyonaka, Ikeda, Suita, Takatsuki, Ibaraki, Minoh, Settsu, Torimoto-cho, Toyono-cho, Nose-cho Tobu Osaka Moriguchi, Hirakata, Yao, Neyagawa, Daito, Kashiwara, Kadoma, Higashi Osaka, Shijonawate, Katano Minami Kawachi Tondabayashi, Kawachinagano, Matsubara, Habikino, Fujiidera, Osaka Sayama, Taishi-cho, Kanan-cho, Chihaya Asaka-mura Senshu Sakai, Kishiwada, Izumiotsu, Kaizuka, Izumisano, Izumi, Takaishi, Sennan, Hannan, Tadaoka-cho, Kumatori-cho, Tajiri-cho, Misaki-cho 8 September 2015 Organization for Academic Affairs Kwansei Gakuin University . -

Goodman Takatsuki
Completion | Mid 2022 Goodman Takatsuki OVERVIEW+ ++ Located inland of Osaka, along Osaka Prefectural road 16 in the Hokusetsu area ++ A modern 4-story logistics facility with a leased area of approximately 6,600 tsubo ++ The surrounding area is densely populated and well-located for employment Driving distance Within 60 minutes driving distance Within 60 minutes Within 30 minutes Kyoto Kyoto Station Nagaokakyo Hyogo Takatsuki + Ibaraki PLANS Goodman Takatsuki Takarazuka Toyonaka Hirakata Amagasaki Nara Osaka StationOsaka A B A Higashi-Osaka Kobe Nara Port Kobe Osaka Airport Port source:Esri and Michael Bauer Research Floor 2/3 Floor 4 LOCATION+ ++ About 2 km from the JR Takatsuki Station and the Hankyu Takatsukishi Station Gross lettable area ( tsubo) ++ A bus stop is located nearby within walking distance Warehouse+ Piloti + About 5.6km from Takatsuki Interchange of Shin-Meishin Expressway, 8km from Ibaraki Interchange of Meishin Floor Office Total + berths driveway Expressway and 8km from Settsu-Kita Interchange of Kinki Expressway 4F A 620 − 40 660 ++ Good access to the Meishin and Shin-Meishin Expressways as well as to the Osaka CBD and Hokusetsu area B 1,275 − 5 1,280 3F A 605 − 130 735 hin-Meishin Expressway 2 km B 1,275 − 5 1,280 2F Takatsuki from JR Takatsuki A B A 605 − 130 735 JCT tation ankyu IC Takatsukishi B 1,050 220 10 1,280 JR Kyoto Line tation 1F Takatsuki A 420 180 50 650 Meishin B 3,600 220 20 3,840 Expressway Hankyu Kyoto Line Total Takatsukishi 5.6 km A 2,250 180 350 2,780 171 from Takatsuki IC Shin-Meishin Expy -
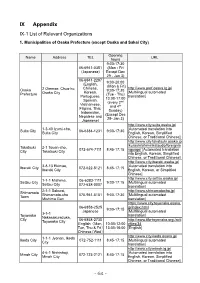
IX Appendix IX-1 List of Relevant Organizations
IX Appendix IX-1 List of Relevant Organizations 1. Municipalities of Osaka Prefecture (except Osaka and Sakai City) Opening Name Address TEL URL hours 9:00-17:30 06-6941-0351 (Mon - Fri (Japanese) Except Dec 29 - Jan 3) 06-6941-2297 9:00-20:00 (English, (Mon & Fri) Chinese, http://www.pref.osaka.lg.jp/ Osaka 2 Otemae, Chuo-ku, 9:00-17:30 Korean, [Multilingual automated Prefecture Osaka City (Tue - Thu) Portuguese, translation] 13:00-17:00 Spanish, (every 2nd Vietnamese, and 4th Filipino, Thai, Sunday) Indonesian, (Except Dec Nepalese and 29- Jan 3) Japanese) http://www.city.suita.osaka.jp/ 1-3-40 Izumi-cho, [Automated translation into Suita City 06-6384-1231 9:00-17:30 Suita City English, Korean, Simplified Chinese, or Traditional Chinese] http://www.city.takatsuki.osaka.jp /kurashi/shiminkatsudo/foreignla Takatsuki 2-1 Touen-cho, 072-674-7111 8:45-17:15 nguage/ [Automated translation City Takatsuki City into English, Korean, Simplified Chinese, or Traditional Chinese] http://www.city.ibaraki.osaka.jp/ 3-8-13 Ekimae, [Automated translation into Ibaraki City 072-622-8121 8:45-17:15 Ibaraki City English, Korean, or Simplified Chinese] http://www.city.settsu.osaka.jp/ 1-1-1 Mishima, 06-6383-1111 Settsu City 9:00-17:15 [Multilingual automated Settsu City 072-638-0007 translation] 2-1-1 Sakurai, http://www.shimamotocho.jp/ Shimamoto Shimamoto-cho 075-961-5151 9:00-17:30 [Multilingual automated Town Mishima Gun translation] https://www.city.toyonaka.osaka. 06-6858-2525 jp/index.html 9:00-17:15 Japanese [Multilingual automated 3-1-1 Toyonaka -

Membership Register MBR0009
LIONS CLUBS INTERNATIONAL CLUB MEMBERSHIP REGISTER SUMMARY THE CLUBS AND MEMBERSHIP FIGURES REFLECT CHANGES AS OF JANUARY 2021 CLUB CLUB LAST MMR FCL YR MEMBERSHI P CHANGES TOTAL DIST IDENT NBR CLUB NAME COUNTRY STATUS RPT DATE OB NEW RENST TRANS DROPS NETCG MEMBERS 5172 023732 ARIDA JAPAN 335 B 4 01-2021 80 1 0 0 -1 0 80 5172 023733 DAITO JAPAN 335 B 4 01-2021 22 0 0 0 0 0 22 5172 023734 FUJIIDERA JAPAN 335 B 4 01-2021 50 3 0 0 0 3 53 5172 023735 HIGASHI OSAKA FUSE JAPAN 335 B 4 01-2021 34 4 0 0 0 4 38 5172 023737 HIGASHI OSAKA KIKUSUI JAPAN 335 B 4 01-2021 38 2 0 1 -2 1 39 5172 023738 GOBO JAPAN 335 B 4 01-2021 42 0 0 0 -1 -1 41 5172 023739 HABIKINO JAPAN 335 B 4 01-2021 50 1 0 0 -1 0 50 5172 023740 HASHIMOTO JAPAN 335 B 4 01-2021 37 2 0 0 -2 0 37 5172 023742 HIRAKATA JAPAN 335 B 4 01-2021 145 4 0 0 -4 0 145 5172 023743 HIGASHI OSAKA JAPAN 335 B 4 01-2021 65 1 0 0 -11 -10 55 5172 023744 IBARAKI JAPAN 335 B 4 01-2021 140 6 0 0 -8 -2 138 5172 023745 IKEDA JAPAN 335 B 4 01-2021 68 1 0 0 -1 0 68 5172 023746 ITO KOYASAN L C JAPAN 335 B 4 01-2021 25 0 0 0 -1 -1 24 5172 023747 IZUMIOSAKA JAPAN 335 B 4 11-2020 34 2 0 0 -1 1 35 5172 023748 IZUMI OTSU JAPAN 335 B 4 01-2021 119 3 0 0 -3 0 119 5172 023750 IZUMISANO CHUO JAPAN 335 B 4 01-2021 46 1 0 0 -2 -1 45 5172 023752 KAINAN JAPAN 335 B 4 01-2021 33 0 0 0 0 0 33 5172 023753 KAIZUKA JAPAN 335 B 4 01-2021 34 1 0 0 0 1 35 5172 023754 KAWACHINAGANO JAPAN 335 B 4 01-2021 29 1 0 0 -2 -1 28 5172 023755 HIGASHI OSAKA KAWACHI JAPAN 335 B 4 08-2020 22 6 0 0 0 6 28 5172 023756 KASHIWARA JAPAN 335 B 4 01-2021 -

Issei Komuro, Department of Cardiovascular Medicine, Osaka University
Appendix The OACIS Investigators Chair: Issei Komuro, Department of Cardiovascular Medicine, Osaka University Graduate School of Medicine, 2-2 Yamada-oka, Suita 565-0871, Japan Secretariats: Yasuhiko Sakata (Chief), Daisaku Nakatani, Shinichiro Suna, Masaya Usami, Sen Matsumoto, Masahiko Hara, Mariko Kishida, Rie Nagai: Department of Cardiovascular Medicine, Osaka University Graduate School of Medicine, Suita, Japan Investigators (institutions listed in alphabetical order): Yoshiyuki Kijima, Yusuke Nakagawa, Minoru Ichikawa, Higashi-Osaka City General Hospital, Higashi-Osaka, Japan; Young-Jae Lim, Shigeo Kawano, Kawachi General Hospital, Higashi-Osaka, Japan; Hiroshi Sato, Kwasnsei Gakuin University, Nishinomiya, Japan; Takashi Shimazu, Hisakazu Fuji, Kazuhiro Aoki, Kobe Ekisaikai Hospital, Kobe, Japan; Seiki Nagata, Yoshio Ishida, Masaaki Uematsu, Tetsuya Watanabe, Masashi Fujita, Masaki Awata, Kansai Rosai Hospital, Amagasaki, Japan; Michio Sugii, Meiwa Hospital, Nishinomiya, Japan; Masatake Fukunami, Takahisa Yamada, Takashi Morita, Osaka General Medical Center, Osaka, Japan; Shinji Hasegawa, Nobuyuki Ogasawara, Osaka Kosei Nenkin Hospital, Osaka, Japan; Tatsuya Sasaki, Yoshinori Yasuoka, Osaka Minami Medical Center, National Hospital Organization, Kawachinagano, Japan; Hideo Kusuoka, Yukihiro Koretsune, Yoshio Yasumura, Keiji Hirooka, Osaka Medical Center, National Hospital Organization, Osaka, Japan; Masatsugu Hori (previous Chair), Osaka Prefectural Hospital Organization Osaka Medical Center for Cancer and Cardiovascular Diseases; -

Summary of Family Membership and Gender by Club MBR0018 As of June, 2009
Summary of Family Membership and Gender by Club MBR0018 as of June, 2009 Club Fam. Unit Fam. Unit Club Ttl. Club Ttl. District Number Club Name HH's 1/2 Dues Females Male TOTAL District 335 B 23732 ARIDA 0 0 0 48 48 District 335 B 23733 DAITO 0 0 0 46 46 District 335 B 23734 FUJIIDERA 0 0 0 26 26 District 335 B 23735 HIGASHI OSAKA FUSE 0 0 3 30 33 District 335 B 23736 HIGASHI OSAKA CHUO 0 0 0 21 21 District 335 B 23737 HIGASHI OSAKA KIKUSUI 0 0 0 38 38 District 335 B 23738 GOBO 0 0 0 51 51 District 335 B 23739 HABIKINO 0 0 0 42 42 District 335 B 23740 HASHIMOTO 0 0 0 32 32 District 335 B 23741 HIGASHI OSAKA APOLLO 0 0 0 20 20 District 335 B 23742 HIRAKATA 0 0 0 73 73 District 335 B 23743 HIGASHI OSAKA 0 0 1 38 39 District 335 B 23744 IBARAKI 0 0 0 92 92 District 335 B 23745 IKEDA 0 0 0 55 55 District 335 B 23746 ITO KOYASAN L C 0 0 0 32 32 District 335 B 23747 IZUMIOSAKA 0 0 0 27 27 District 335 B 23748 IZUMI OTSU 0 0 0 69 69 District 335 B 23749 IZUMISANO 0 0 0 26 26 District 335 B 23750 IZUMISANO CHUO 0 0 0 35 35 District 335 B 23751 KADOMA 0 0 0 20 20 District 335 B 23752 KAINAN 0 0 0 30 30 District 335 B 23753 KAIZUKA 0 0 0 34 34 District 335 B 23754 KAWACHINAGANO 1 0 2 34 36 District 335 B 23755 HIGASHI OSAKA KAWACHI 0 0 2 25 27 District 335 B 23756 KASHIWARA 0 0 0 68 68 District 335 B 23758 KATSUURA 0 0 0 23 23 District 335 B 23759 KISHIWADA CHIKIRI 0 0 0 44 44 District 335 B 23760 KISHIWADA 0 0 0 45 45 District 335 B 23761 KISHIWADA CHUO 0 0 2 55 57 District 335 B 23763 KONGO 1 1 0 28 28 District 335 B 23764 KUSHIMOTO 0 0 2 24 -

IX-3 Health and Medical Care
IX-3 Health and Medical Care 1. Emergency Medical Clinics (Service available only in Japanese. It is recommended that you bring along someone competent in Japanese.) Int Internal Ped Pediatrics Medicine Sur General Den Dentistry Surgery Oph Ophthalmology Oto Otolaryngology Ort Orthopedics ※ You can find more information using Osaka Medical Facilities Information System search enging. (https://www.mfis.pref.osaka.jp/apqq/qq/men/pwtpmenult01.aspx) Town Facility Name Address Tel Reception Hours Suita Municipal 19-2 Deguchi-cho, Sun, Holidays, Year-end Suita Emergency Clinic Suita 06-6339-2271 & New Year Int Ped Sur Den 9:30-11:30, 13:00-16:30 Weeknights (Int・Ped・Sur)20:30-6:30 Osaka Mishima Sat (Int・Ped・Sur) Emergency 14:30-6:30 11-1 Medical Center/ Sun and Holidays Minami-akutagawa Shimamoto & Takatsuki 072-683-9999 cho, Takatsuki (Int・Ped・Sur) Takatsuki Shimamoto 9:30-11:30, 13:30-16:30, Emergency Clinic 18:30-6:30 Int Ped Sur Den Sun and Holidays (Den) 9:30-11:30, 13:30-16:30 Weeknights Ibaraki Municipal (Int) 21:00-23:30 Public Health and Sat (Int) 17:00-6:30 Medical Center Emergency Clinic Sun and Holidays 3-13-5 Kasuga, Ibaraki Int. Den 072-625-7799 (Int) Ibaraki 10:00-11:30, 13:00-16:30, *For pediatrics, 18:00-6:30 visit Takatsuki/ Sun and Holidays Shimamoto Emergency Clinic (Den) 10:00-11:30, 13:00-16:30 Settsu Municipal Sun, Holidays, Year-end Emergency 32-19 Kohroen, Settsu 072-633-1171 & New Year Pediatric Clinic Settsu Ped 10:00-11:30, 13:30-16:30 Clinic of Toyonaka Sun, Holidays, Aug 14/15, Municipal Health 2-6-1 Uenosaka, 06-6848-1661 -
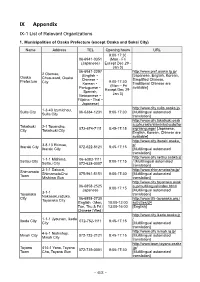
IX Appendix IX-1 List of Relevant Organizations
IX Appendix IX-1 List of Relevant Organizations 1. Municipalities of Osaka Prefecture (except Osaka and Sakai City) Name Address TEL Opening hours URL 9:00-17:30 06-6941-0351 (Mon - Fri (Japanese) Except Dec 29 - Jan 3) 06-6941-2297 http://www.pref.osaka.lg.jp/ 2 Otemae, (English・ [Japanese, English, Korean, Osaka Chuo-ward, Osaka Chinese・ Simplified Chinese, Prefecture 9:00-17:30 City Korean・ Traditional Chinese are (Mon – Fri Portuguese・ available] Except Dec 29- Spanish, Jan 3) Vietnamese・ Filipino・Thai・ Japanese) http://www.city.suita.osaka.jp/ 1-3-40 Izumichou, Suita City 06-6384-1231 9:00-17:30 [Multilingual automated Suita City translation] http://www.city.takatsuki.osak a.jp/kurashi/shiminkatsudo/for Takatsuki 2-1 Touencho, 072-674-7111 8:45-17:15 eignlanguage/ [Japanese, City Takatsuki City English, Korean, Chinese are available] http://www.city.ibaraki.osaka.j 3-8-13 Ekimae, p/ Ibaraki City 072-622-8121 8:45-17:15 Ibaraki City [Multilingual automated translation] http://www.city.settsu.osaka.jp 1-1-1 Mishima, 06-6383-1111 Settsu City 9:00-17:15 / [Multilingual automated Settsu City 072-638-0007 translation] 2-1-1 Sakurai, http://www.shimamotocho.jp/ Shimamoto ShimamotoCho 075-961-5151 9:00-17:30 [Multilingual automated Town Mishima Gun translation] http://www.city.toyonaka.osak 06-6858-2525 a.jp/multilingual/index.html/ 9:00-17:15 Japanese [Multilingual automated 3-1-1 Toyonaka translation] Nakasakurazuka, City 06-6858-2730 http://www.tifa-toyonaka.org./ Toyonaka City English(Mon, 10:00-12:00 activities/34 Tue, Thu & Fri) 13:00-16:00 -

A Case of Lake Biwa - Yodo River System
Watershed Management in Drinking Water for Emergency: A Case of Lake Biwa - Yodo River System Takashi SASAKI*1& Daiji NAGASHIO** Yodo River Water Quality Committee 1. Background Lake Biwa - Yodo River water system is located in the middle west part of Japan and supports urban life and activities as one of the main water source for 14 million consumers in the Kansai area (Fig. 1). The water system is characterized by urbanization and developed industry in upstream area, at the same time, much use of domestic water in downstream area. This means the water is being reused so highly from upstream to downstream that there exist many kinds of risk factors to water safety. Drinking water supply utilities taking water from the river organize “Yodo River Water Quality Committee” for integrated source water quality management to cope quickly and systematically with source accidents. This paper describes outline of the Committee and its source monitoring for emergency. 2. Outline of Yodo River Water Quality Committee Fig. 2 shows yearly changes in raw water quality at Kunijima water treatment plant (WTP) of Osaka City, one of the largest utilities. Since the quality rapidly deteriorated due to increased domestic and industrial wastewaters in 1960s, downstream utilities were forced to take countermeasures for these problems. The utilities concluded that cooperative approach for integrated source quality management was the best way and established Yodo River Water Quality Committee in 1965, which is currently composed of ten utilities (Osaka Prefectural -, Osaka Municipal -, Moriguchi City -, Neyagawa City -, Hirakata City -, Suita City -, Amagasaki City -, Itami City -, Nishinomiya City - waterworks and Hanshin Water Supply Authority) (Pic. -

Membership Register MBR0009
LIONS CLUBS INTERNATIONAL CLUB MEMBERSHIP REGISTER SUMMARY THE CLUBS AND MEMBERSHIP FIGURES REFLECT CHANGES AS OF NOVEMBER 2019 CLUB CLUB LAST MMR FCL YR MEMBERSHI P CHANGES TOTAL DIST IDENT NBR CLUB NAME COUNTRY STATUS RPT DATE OB NEW RENST TRANS DROPS NETCG MEMBERS 5172 023732 ARIDA JAPAN 335 B 4 11-2019 84 0 0 0 0 0 84 5172 023733 DAITO JAPAN 335 B 4 11-2019 21 1 0 0 0 1 22 5172 023734 FUJIIDERA JAPAN 335 B 4 11-2019 47 2 0 0 -1 1 48 5172 023735 HIGASHI OSAKA FUSE JAPAN 335 B 4 10-2019 33 2 0 0 0 2 35 5172 023737 HIGASHI OSAKA KIKUSUI JAPAN 335 B 4 11-2019 40 2 0 0 0 2 42 5172 023738 GOBO JAPAN 335 B 4 11-2019 38 3 0 0 -1 2 40 5172 023739 HABIKINO JAPAN 335 B 4 11-2019 50 2 0 0 -2 0 50 5172 023740 HASHIMOTO JAPAN 335 B 4 11-2019 36 1 0 0 -2 -1 35 5172 023742 HIRAKATA JAPAN 335 B 4 11-2019 118 5 0 1 -4 2 120 5172 023743 HIGASHI OSAKA JAPAN 335 B 4 11-2019 58 0 0 0 -2 -2 56 5172 023744 IBARAKI JAPAN 335 B 4 10-2019 130 11 0 0 0 11 141 5172 023745 IKEDA JAPAN 335 B 4 11-2019 55 3 0 0 -1 2 57 5172 023746 ITO KOYASAN L C JAPAN 335 B 4 11-2019 24 0 0 0 0 0 24 5172 023747 IZUMIOSAKA JAPAN 335 B 4 10-2019 22 5 0 0 0 5 27 5172 023748 IZUMI OTSU JAPAN 335 B 4 11-2019 101 10 0 0 -1 9 110 5172 023750 IZUMISANO CHUO JAPAN 335 B 4 11-2019 39 2 0 0 -2 0 39 5172 023752 KAINAN JAPAN 335 B 4 11-2019 31 1 0 0 0 1 32 5172 023753 KAIZUKA JAPAN 335 B 4 10-2019 36 0 0 1 0 1 37 5172 023754 KAWACHINAGANO JAPAN 335 B 4 10-2019 30 1 0 0 0 1 31 5172 023755 HIGASHI OSAKA KAWACHI JAPAN 335 B 4 11-2019 29 2 0 0 0 2 31 5172 023756 KASHIWARA JAPAN 335 B 4 11-2019