Errors of Morphogenesis and Developmental Field Theory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download PDF File
Folia Morphol. Vol. 77, No. 2, pp. 386–392 DOI: 10.5603/FM.a2017.0080 O R I G I N A L A R T I C L E Copyright © 2018 Via Medica ISSN 0015–5659 www.fm.viamedica.pl Incidental imaging findings of congenital rib abnormalities: a case series and review of developmental concepts A.M. Aignătoaei1, C.E. Moldoveanu2, I.-D. Căruntu3, S.E. Giuşcă3, S. Partene Vicoleanu3, A.H. Nedelcu3 1Department of Pathology, “Sf. Spiridon” Clinic Hospital, 1, Independenţei Square, Iaşi, Romania 2Department of Radiology, Pneumology Clinic Hospital, 30, dr. Iosif Cihac Street, Iaşi, Romania 3Department of Morphofunctional Sciences, “Grigore T. Popa” University, 16, University Street, Iaşi, Romania [Received: 12 June 2017; Accepted: 24 July 2017] Background: Congenital rib abnormalities are found in approximately 2% of the general population. Usually, they occur in isolation and are rarely symptomatic, but they can also be associated with other malformations. Materials and methods: We reviewed imaging examinations performed over a period of 2 years (2014–2015), enabling us to identify isolated rib abnormalities in 6 adult patients. Results: The case series consisted in 3 cases with bilateral cervical ribs and 1 case each with bifid rib, costal fusion and rib pseudarthrosis. In all patients, the costal anomalies were discovered incidentally. All rib malformations were detected at thoracic radiography, except for the rib pseudarthrosis, which was identified at computed tomography (CT) scan. Differential diagnosis was made between cer- vical ribs and abnormalities of the C7 transverse process and of the first rib, while the other costal malformations were distinguished from tumoural, traumatic or inflammatory lesions of the chest wall, lung and pleura. -

Modeling Congenital Disease and Inborn Errors of Development in Drosophila Melanogaster Matthew J
© 2016. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2016) 9, 253-269 doi:10.1242/dmm.023564 REVIEW SUBJECT COLLECTION: TRANSLATIONAL IMPACT OF DROSOPHILA Modeling congenital disease and inborn errors of development in Drosophila melanogaster Matthew J. Moulton and Anthea Letsou* ABSTRACT particularly difficult to manage clinically {e.g. CHARGE syndrome Fly models that faithfully recapitulate various aspects of human manifesting coloboma [emboldened words and phrases are defined disease and human health-related biology are being used for in the glossary (see Box 1)], heart defects, choanal atresia, growth research into disease diagnosis and prevention. Established and retardation, genitourinary malformation and ear abnormalities new genetic strategies in Drosophila have yielded numerous (Hsu et al., 2014), and velocardiofacial or Shprinstzens syndrome substantial successes in modeling congenital disorders or inborn manifesting cardiac anomaly, velopharyngeal insufficiency, errors of human development, as well as neurodegenerative disease aberrant calcium metabolism and immune dysfunction (Chinnadurai and cancer. Moreover, although our ability to generate sequence and Goudy, 2012)}. Estimates suggest that the cause of at least 50% of datasets continues to outpace our ability to analyze these datasets, congenital abnormalities remains unknown (Lobo and Zhaurova, the development of high-throughput analysis platforms in Drosophila 2008). It is vital that we understand the etiology of congenital has provided access through the bottleneck in the identification of anomalies because this knowledge provides a foundation for improved disease gene candidates. In this Review, we describe both the diagnostics as well as the design of preventatives and therapeutics that traditional and newer methods that are facilitating the incorporation of can effectively alleviate or abolish the effects of disease. -

Medical Genetics and Genomic Medicine in the United States of America
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by George Washington University: Health Sciences Research Commons (HSRC) Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 7-1-2017 Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease. Carlos R Ferreira George Washington University Debra S Regier George Washington University Donald W Hadley P Suzanne Hart Maximilian Muenke Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Genetics and Genomics Commons APA Citation Ferreira, C., Regier, D., Hadley, D., Hart, P., & Muenke, M. (2017). Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease.. Molecular Genetics and Genomic Medicine, 5 (4). http://dx.doi.org/10.1002/mgg3.318 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. GENETICS AND GENOMIC MEDICINE AROUND THE WORLD Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease Carlos R. Ferreira1,2 , Debra S. Regier2, Donald W. Hadley1, P. Suzanne Hart1 & Maximilian Muenke1 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 2Rare Disease Institute, Children’s National Health System, Washington, District of Columbia Correspondence Carlos R. -

Part II Thoracic Spine
Open Access Review Article DOI: 10.7759/cureus.8684 Anatomical Variations That Can Lead to Spine Surgery at The Wrong Level: Part II Thoracic Spine Manan Shah 1 , Dia R. Halalmeh 2 , Aubin Sandio 1 , R. Shane Tubbs 3, 4, 5 , Marc D. Moisi 2 1. Neurosurgery, Wayne State University, Detroit Medical Center, Detroit, USA 2. Neurosurgery, Detroit Medical Center, Detroit, USA 3. Neurosurgery and Structural & Cellular Biology, Tulane University School of Medicine, New Orleans, USA 4. Anatomical Sciences, St. George's University, St. George's, GRD 5. Neurosurgery and Ochsner Neuroscience Institute, Ochsner Health System, New Orleans, USA Corresponding author: Dia R. Halalmeh, [email protected] Abstract Spine surgery at the wrong level is a detrimental ordeal for both surgeon and patient, and it falls under the wrong-site surgery sentinel events reporting system. While there are several methods designed to limit the incidence of these events, they continue to occur and can result in significant morbidity for the patient and malpractice lawsuits for the surgeon. In thoracic spine, numerous risk factors influence the development of this misadventure. These include anatomical variations such as transitional vertebrae, rib variants, hemivertebra, and block/fused vertebrae as well as patient characteristics, such as tumors, infections, previous thoracic spine surgery, obesity, and osteoporosis. An extensive literature search of the PubMed database up to 2019 was completed on each of the anatomical entities and their influence on developing thoracic spine surgery at the wrong level, taking into consideration patient’s individual factors. A reliable protocol and effective techniques were described to prevent this error. -

Nova Scotia Atlee Perinatal Database Coding Manual 8Th Edition (Version 8.0)
Nova Scotia Atlee Perinatal Database Coding Manual 8th Edition (Version 8.0) April 2001 NOVA SCOTIA ATLEE PERINATAL DATABASE CODING MANUAL TABLE OF CONTENTS (A) ROUTINE INFORMATION LISTING OF HOSPITALS .......................1 DELIVERED ADMISSIONS ......................3 UNDELIVERED ADMISSIONS ..................59 POSTPARTUM ADMISSIONS ...................69 NEONATAL ADMISSIONS .................... 79 ANOMALY ADMISSIONS ......................91 ................................................. (B) CLASSIFICATION OF MATERNAL DISEASES AND PROCEDURES (Blue Section) I. Previous Pregnancy Maternal Diseases.............. 01 II. Present Pregnancy Maternal Diseases............... 02 A. Obstetrical ..............................02 B. Non-Obstetrical..........................07 C. Labour Complications .....................15 D. Non-Delivery Procedures ..................16 E. Analgesia During Labour ...................20 F. Anesthesia During Labour/Delivery ..........22 G. Anesthesia for Non-Delivery Procedures ......24 H. Lacerations ..............................25 I. Postpartum Complications..................26 J. Postpartum Infections .....................29 K. Maternal Therapy.........................31 L. Maternal Death or Undelivered Fetal Death....33 M. Infection in Present Pregnancy .............34 N. Complications of Anesthesia................37 O. Antibiotic Therapy........................39 P. Fetal Procedures..........................40 Q. Methods of Induction ......................41 8th Edition, April 2001, V8.0 Table of Contents and -

Jaw Keratocysts and Sotos Syndrome
International Journal on Oral Health Jaw Keratocysts and Sotos Syndrome Case Report Volume 1 Issue 2- 2021 Author Details Jin Fei Yeo and Philip McLoughlin* Faculty of Dentistry, National University Centre for Oral Health Singapore, Singapore *Corresponding author Philip McLoughlin, Discipline of Oral & Maxillofacial Surgery, Faculty of Dentistry, National University Centre for Oral Health Singapore, 9 Lower Kent Ridge Road, Singapore Article History Received: June 05, 2021 Accepted: June 14, 2021 Published: June 15, 2021 Abstract Sotos syndrome, described by Sotos et al. [1], is characterized by excessive growth during childhood, macrocephaly, distinctive facial appearance and learning disability. The disorder is largely caused by mutations or deletions in the NSD1 gene. The typical facial gestalt includes macrodolichocephaly with frontal bossing, front-parietal sparseness of hair, apparent hypertelorism, down slanting palpebral in his jaw bones, a previously unreported oral manifestation, out with a syndromic context. fissures, and facial flushing. This paper discusses a case of Sotos syndrome in an adolescent male with multiple odontogenic keratocysts Introduction On examination, he was a generally healthy and rather active patient. He was very tall with macrocephaly, large hands and feet, and Sotos syndrome (Cerebral gigantism) is a rare genetic childhood hypertelorism with slightly down-slanted eyes. He also suffered ASD/ overgrowth condition described in 1964 by Sotos et al. [1]. This autism and mild mental hypo development with noticeable speech syndrome is characterized by excessive growth during childhood, impairment. On clinical assessment he was found to have a number of macrocephaly, distinctive facial appearance and learning disability. unerupted teeth, with fluctuant expansion of the maxilla and mandible The disorder is caused by mutations or 5q35 microdeletions in the at those sites. -

Medical Genetics and Genomic Medicine in the United States of America
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 7-1-2017 Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease. Carlos R Ferreira George Washington University Debra S Regier George Washington University Donald W Hadley P Suzanne Hart Maximilian Muenke Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Genetics and Genomics Commons APA Citation Ferreira, C., Regier, D., Hadley, D., Hart, P., & Muenke, M. (2017). Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease.. Molecular Genetics and Genomic Medicine, 5 (4). http://dx.doi.org/10.1002/mgg3.318 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. GENETICS AND GENOMIC MEDICINE AROUND THE WORLD Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease Carlos R. Ferreira1,2 , Debra S. Regier2, Donald W. Hadley1, P. Suzanne Hart1 & Maximilian Muenke1 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 2Rare Disease Institute, Children’s National Health System, Washington, District of Columbia Correspondence Carlos R. Ferreira, National Human Genome Research Institute, National Institutes of Health, 10 Center Drive, Building 10, Room 10C103, Bethesda, Maryland 20892-1851. -

PTCH1 Duplication in a Family with Microcephaly and Mild Developmental Delay
European Journal of Human Genetics (2009) 17, 267 – 271 & 2009 Macmillan Publishers Limited All rights reserved 1018-4813/09 $32.00 www.nature.com/ejhg SHORT REPORT PTCH1 duplication in a family with microcephaly and mild developmental delay Katarzyna Derwin´ ska1,2, Marta Smyk1,2, Mitchell Lance Cooper1, Patricia Bader3, Sau Wai Cheung1 and Paweł Stankiewicz*,1 1Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; 2Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland; 3Department of Cytogenetics, Parkview Memorial Hospital, Fort Wayne, IN, USA With the exception of the X chromosome, genomic deletions appear to be more prevalent than duplications. Because of a lack of accurate diagnostic methods, submicroscopic duplications have been under-ascertained for a long period. The development of array CGH has enabled the detection of chromosomal microduplications with nearly the same sensitivity as deletions, leading to the discovery of previously unrecognized syndromes. Using a clinical targeted oligonucleotide array (CMA-V6.3 OLIGO), we identified an B360-kb duplication in 9q22.32 in a 21-month-old boy with developmental delay, failure to thrive, and microcephaly. The same duplication was identified in the patient’s mother who is also microcephalic and mildly delayed. We have sequenced the chromosomal breakpoints and determined the duplication as tandem in orientation and 363 599 bp in size. The duplicated segment harbors the entire PTCH1 gene. Deletions or loss-of-function mutations of PTCH1 result in basal cell nevus syndrome (Gorlin syndrome), whereas gain-of-function mutations were proposed to lead to holoprosencephaly 7. We propose that patients with microcephaly or holoprosencephaly of unknown origin should also be screened for PTCH1 duplication. -
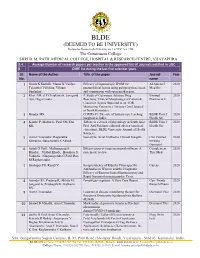
BLDE(Deemed to Be University)
BLDE (DEEMED TO BE UNIVERSITY) Declared as Deemed to be University u/s 3 of UGC Act, 1956 The Constituent College SHRI B. M. PATIL MEDICAL COLLEGE, HOSPITAL & RESEARCH CENTRE, VIJAYAPURA 3.4. Average Number of research papers per teacher in the approved list of Journals notified in UGC- 6 CARE list during the last five calendar years Sl. Name of the Author Title of the paper Journal Year No. name 1 Girish K Kullolli, Manoj K Vaidya, Efficacy of laparoscopic IPOM for Al Ameen J 2020 Tejaswini Vallabha, Vikram paraumbilical hernia using polypropylene mesh Med Sci Sindgikar. and comparison with open mesh repair. 2 Khot AM, S P Chaukimath, Janagond A Study of Cutaneous Adverse Drug Biomed 2020 Ajit, Hugar Leela. Reactions; Clinical/Morphological Pattern & Pharmacol J Causative Agents Reported in an ADR Monitoring Centre in a Tertiary Care Hospital of North Karnataka. 3 Biradar MS. COVID-19: The role of tertiary care teaching BLDE Univ J 2020 hospitals in India. Health Sci 4 Kanthe P, Mullur L, Patil SM, Das Tribute to a doyen of physiology in South Asia: BLDE Univ J 2020 KK. Prof. Arif Siddiqui, editorial adviser (medical Health Sci education), BLDE University Journal of Health Sciences. 5 Arun C Inamadar, Ragunatha Necrolytic Acral Erythema: Current Insights Clin Cosmet 2020 Shivanna, Balachandra S Ankad. Investig Dermatol 6 Satish G Patil , Mallanagoud S Effectiveness of yoga on arterial stiffness: A Complement 2020 Biradar, Vitthal Khode, Hosakote S systematic review Ther Med Vadiraja, Ninganagouda G Patil, Rao M Raghavendra. 7 Shahapur PR, Kandi V. Seroprevalence of Rubella Virus-specific Cureus 2020 Antibodies in Women and the Diagnostic Efficacy of Enzyme-linked Immunoassay and Rapid Immunochromatographic Tests. -

Methotrexate
Ó 2012 Wiley Periodicals, Inc. Birth Defects Research (Part A) 00:000–000 (2012) Teratogen Update: Methotrexate Sara C. Hyoun,1 Sarah G. Obicˇan,2 and Anthony R. Scialli2,3* 1George Washington University, School of Medicine and Health Sciences, Washington, D.C 2Department of Obstetrics and Gynecology, George Washington University Medical Center and Reproductive Toxicology Center, Washington, D.C 3Tetra Tech Sciences, Arlington, Virginia Received 9 December 2011; Revised 9 January 2012; Accepted 13 January 2012 Methotrexate and aminopterin are folic acid antagonists that inhibit dihydrofolate reductase, resulting in a block in the synthesis of thymidine and inhibition of DNA synthesis. Methotrexate has been used for the treatment of malignancy, rheumatic disorders, and psoriasis and termination of intrauterine pregnancy. Recently, methotrexate has become a standard treatment for ectopic pregnancy. The misdiagnosis of an intrauterine pregnancy as an ectopic pregnancy can result in exposure of a continuing pregnancy to dose levels of methotrexate of 50 mg/m2 (maternal body surface area). Experimental animal studies have associ- ated methotrexate therapy with embryo death in mice, rats, rabbits, and monkeys. Structural malformations have been most consistently produced in rabbits at a maternal dose level of 19.2 mg/kg. Abnormalities in rabbits include hydrocephalus, microphthalmia, cleft lip and palate, micrognathia, dysplastic sacral and cau- dal vertebrate, phocomelia, hemimelia, syndactyly, and ectrodactyly. Based on human case reports of metho- trexate exposure during pregnancy, a methotrexate embryopathy has been described that includes growth deficiency, microcephaly, hypoplasia of skull bones, wide fontanels, coronal or lambdoidal craniosynostosis, upswept frontal scalp hair, broad nasal bridge, shallow supraorbital ridges, prominent eyes, low-set ears, maxillary hypoplasia, epicanthal folds, short limbs, talipes, hypodactyly, and syndactyly. -

Scholars Journal of Dental Sciences Oral Manifestations of Some Rare
Scholars Journal of Dental Sciences Abbreviated Key Title: Sch J Dent Sci ISSN 2394-4951 (Print) | ISSN 2394-496X (Online) Journal homepage: https://saspublishers.com/sjds/ Oral Manifestations of Some Rare Diseases: A Narrative Review of the Radiological Findings in Orthopantomography Luca Viganò1*, Cinzia Casu2, Mariapia Brignoglio3, Veronica Caria3, Marina Paola Lazzari4, Clarita Pellegrini3 1Department of Radiology, San Paolo Dental Building, University of Milan, Italy 2Private Dental Practice, Cagliari, Italy 3San Paolo Dental Building, University of Milan, Italy 4IRCCS Ca’ Granda Foundation General Hospital University of Milan, Italy DOI: 10.36347/sjds.2020.v07i10.003 | Received: 28.09.2020 | Accepted: 08.10.2020 | Published: 15.10.2020 *Corresponding author: Luca Viganò Abstract Original Research Article Rare diseases have a prevalence that is not more than 5 cases per 10,000 people. Some rare diseases have oral manifestations. Among these conditions, there are some syndromes, such as Gorlin Goltz, Turner syndrome, SAPHO, Williams syndrome, Schimke Immuno, Cherubism, and Neurofibromatosis type 1. Considering their low incidence and high complexity, these disorders require a diagnostic and therapeutic approach based on the interaction of several specialists. In these field, the dentist plays a leading role: he is at the forefront of the early detection of diseases whose prodromal signs may forerun systemic manifestations by few years. Orthopantomography (OPT) allows to identify changes in bone structures and dental anomalies of shape and number, often associated with the above listed manifestations. The aim of this article is to show how the dentist, basing on some radiological evidences, may be able to make an early diagnosis or to formulate a diagnostic hypothesis that will need to be approved by a multidisciplinary team. -

Living History-Biography: from Oral Pathology to Craniofacial Genetics
American Journal of Medical Genetics 46:317-334 (1993) Living History-Biography: From Oral Pathology to Craniofacial Genetics Robert J. Gorlin University of Minnesota School of Dentistry, Minneapolis, Minnesota ROBERT JAMES GORLIN Early Life I was born on 11 January 1923 in Hudson, N.Y.,the only child of James Alter Gorlin and Gladys Gretchen Hallenbeck. Abandoned by my mother, my father placed me in the care of my great aunt who raised me until I was 11 years old when she began to suffer ill health and had no more economic wherewithal to support me fur- ther. I then joined my father and my stepmother whom he had recently married. My father was a small scale businessman with little formal education. We moved several times within a few years to small towns in New Jersey, partly the result of his poor business acumen and partly due to malencounters with the law. In 1937, we moved to Newark and settled in a two-bedroom apart- ment with grandparents, uncles, aunts, cousins, and two boarders. In spite of the crowded conditions, every niche and recess being occupied by a bed, it was not, to the best of my memory, an unhappy home. I do not know what hopes or ambitions my father and stepmother had for me. They respected education but they believed that since they were so impecunious, col- lege was out of the picture. After I rejected the idea of becoming a priest (my stepmother’s brother’s calling), my father suggested that I take a General Business Preparation curriculum in high school.