Geospatial Resolution of Human and Bacterial Diversity with City-Scale Metagenomics Ebrahim Afshinnekoo CUNY Queens College
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Manuscript III
Manuscript III 60 Manuscript Initial characterization of ParI, an orphan C5-DNA methyltransferase from Psychrobacter arcticus 273-4 Miriam Grgic1, Gro Elin Kjæreng Bjerga2, Adele Kim Williamson1, Bjørn Altermark1 and Ingar Leiros1 1Department of chemistry, University of Tromsø, The Arctic University of Norway, Tromsø, Norway 2 University of Bergen, Bergen, Norway 1 Abstract Background DNA methyltransferase are important enzymes in most living organisms, from single cell bacteria to eukaryotes. In prokaryotes, most DNA methyltransferases are members of the bacterial type II restriction modification system: The DNA methyltransferases methylates host DNA, thereby protecting it from cleavage by the restriction endonucleases that recognize the same sequence. In addition to being part of restriction modification systems, DNA methyltransferases can act as orphan enzymes having important role in controlling gene expression, DNA replication, cell cycle and DNA post replicative mismatch repair. In higher eukaryotes, DNA methylation is involved in the regulation of gene expression, maintenance of genome integrity, parental imprinting, chromatin condensation as well as being involved in development of diseases and disorders. DNA-MTases have several potential applications in biotechnology for example in targeted gene silencing and in DNA-protein interaction studies Results According to bioinformatical analyses, the parI gene from Psychrobacter arcticus 273-4, encoding a cytosine specific DNA methyltransferase, is likely of phage origin. In this work, ParI was expressed and purified in a McrBC negative E. coli strain in fusion with a hexahistidine affinity tag and maltose binding protein solubility protein. The obtained recombinant ParI was monomeric with a molecular mass estimated to 54 kDa. The melting temperature of the protein was determined to be 53 ºC in ThermoFlour assay and 54 ºC in differential scanning calorimetry, with no secondary structures detectable at 65 ºC. -
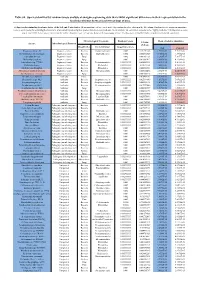
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Characterization and Biological Activity of Bacterial Glycoconjugates in Cold Adaptation
UNIVERSITA’ DEGLI STUDI DI NAPOLI FEDERICO II Chemical Science XXVIII Cycle Characterization and biological activity of bacterial glycoconjugates in cold adaptation. Angela Casillo Tutor: Prof. Maria Michela Corsaro Supervisor: Prof. Angela Amoresano 1 Summary Abbreviation 6 Abstract 8 Introduction Chapter I: Microorganisms at the limits of life 17 1.1 Psychrophiles 1.2 Molecular and physiological adaptation 1.3 Industrial and biotechnological applications Chapter II: Gram-negative bacteria 25 2.1 Gram-negative cell membrane: Lipopolysaccharide 2.2 Bacterial extracellular polysaccharides (CPSs and EPSs) 2.3 Biofilm 2.4 Polyhydroxyalkanoates (PHAs Chapter III: Methodology 36 3.1 Extraction and purification of LPS 3.2 Chemical analysis and reactions on LPS 3.2.1 Lipid A structure determination 3.2.2 Core region determination 3.3 Chromatography in the study of oligo/polysaccharides 3.4 Mass spectrometry of oligo/polysaccharides 3.5 Nuclear Magnetic Resonance (NMR) Results Colwellia psychrerythraea strain 34H Chapter IV: C. psychrerythraea grown at 4°C 47 4.1 Lipopolysaccharide and lipid A structures 4.1.1Isolation and purification of lipid A 4.1.2 ESI FT-ICR mass spectrometric analysis of lipid A 4.1.3 Discussion 4.2 Capsular polysaccharide (CPS) 4.2.1 Isolation and purification of CPS 4.2.2 NMR analysis of purified CPS 4.2.3 Three-dimensional structure characterization 4.2.4 Ice recrystallization inhibition assay 4.2.5 Discussion 4.3 Extracellular polysaccharide (EPS) 4.3.1 Isolation and purification of EPS 4.3.2 NMR analysis of purified EPS 4.3.3 Three-Dimensional Structure Characterization 4.3.4 Ice Recrystallization Inhibition assay 4.3.5 Discussion 4.4 Mannan polysaccharide 4.4.1 Isolation and purification of Mannan polysaccharide 4.4.2 NMR analysis 4.4.3 Ice Recrystallization Inhibition assay 4.4.4 Discussion 4.5 Polyhydroxyalkanoates (PHA) 4.5.1 Discussion Conclusion Chapter V: C. -

Marine Drugs
Mar. Drugs 2015, 13, 4539-4555; doi:10.3390/md13074539 OPEN ACCESS marine drugs ISSN 1660-3397 www.mdpi.com/journal/marinedrugs Article Structural Investigation of the Oligosaccharide Portion Isolated from the Lipooligosaccharide of the Permafrost Psychrophile Psychrobacter arcticus 273-4 Angela Casillo 1, Ermenegilda Parrilli 1, Sannino Filomena 1,2, Buko Lindner 3, Rosa Lanzetta 1, Michelangelo Parrilli 4, Maria Luisa Tutino 1 and Maria Michela Corsaro 1,* 1 Dipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Complesso Universitario Monte S. Angelo, Via Cintia 4, Napoli 80126, Italy; E-Mails: [email protected] (A.C.); [email protected] (E.P.); [email protected] (S.F.); [email protected] (R.L.); [email protected] (M.L.T.) 2 Institute of Protein Biochemistry, CNR, Via Pietro Castellino 111, Napoli 80131, Italy 3 Division of Bioanalytical Chemistry, Research Center Borstel, Leibniz-Center for Medicine and Biosciences, Parkallee 10, BorstelD-23845, Germany; E-Mail: [email protected] 4 Dipartimento di Biologia, Università degli Studi di Napoli Federico II, Complesso Universitario Monte S. Angelo, Via Cintia 4, Napoli 80126, Italy; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-081-674149; Fax: +39-081-674393. Academic Editor: Antonio Trincone Received: 22 June 2015 / Accepted: 14 July 2015 /Published: 22 July 2015 Abstract: Psychrophilic microorganisms have successfully colonized all permanently cold environments from the deep sea to mountain and polar regions. The ability of an organism to survive and grow in cryoenviroments depends on a number of adaptive strategies aimed at maintaining vital cellular functions at subzero temperatures, which include the structural modifications of the membrane. -

Biochemical and Molecular Characterization of Putative Immunoprotective Molecules of the Soft Tick, Ornithodoros Savignyi Audouin (1827)
Biochemical and molecular characterization of putative immunoprotective molecules of the soft tick, Ornithodoros savignyi Audouin (1827) By Po Hsun (Paul) Cheng Submitted in partial fulfillment of the degree Philosophiae Doctor Department of Biochemistry In the Faculty of Natural and Agricultural Sciences University of Pretoria Pretoria November 2010 © University of Pretoria Table of contents Page List of Figures IV List of Tables VI Abbreviations VII Acknowledgements X Summary XII Chapter: 1 Introduction 1 1.1 Problem statement 1 1.2 Ornithodoros savignyi as a model organism for this study 2 1.3 The invertebrate innate immunity 3 1.3.1 Discrimination between self and non-self 4 1.3.2 Cellular immune responses 7 1.3.2.1 Phagocytosis 8 1.3.2.2 Nodulation and encapsulation 9 1.3.2.3 Melanization 10 1.3.3 Humoral immune responses 11 1.3.3.1 AMPs 11 1.3.3.2 Lysozyme 15 1.3.3.3 Proteases and protease inhibitors 18 1.3.4 Coagulation 19 1.3.5 Iron sequestration 19 1.3.6 Oxidative stress 20 1.4 Aims of thesis 21 Chapter 2: Identification of tick proteins recognizing Gram-negative 22 bacteria 2.1 Introduction 22 2.2 Hypothesis 24 2.3 Materials and methods 25 2.3.1 Ticks 25 2.3.2 Reagents 25 2.3.3 E. coli binding proteins in hemolymph plasma from unchallenged 26 ticks 2.3.3.1 Binding assay 26 2.3.3.2 SDS-PAGE analysis of bacteria binding proteins in plasma from 27 unchallenged ticks 2.3.4 E. -

Psychrophiles
EA41CH05-Cavicchioli ARI 30 April 2013 11:9 Psychrophiles Khawar S. Siddiqui,1 Timothy J. Williams,1 David Wilkins,1 Sheree Yau,1 Michelle A. Allen,1 Mark V. Brown,1,2 Federico M. Lauro,1 and Ricardo Cavicchioli1 1School of Biotechnology and Biomolecular Sciences and 2Evolution and Ecology Research Center, The University of New South Wales, Sydney, New South Wales 2052, Australia; email: [email protected] Annu. Rev. Earth Planet. Sci. 2013. 41:87–115 Keywords First published online as a Review in Advance on microbial cold adaptation, cold-active enzymes, metagenomics, microbial February 14, 2013 diversity, Antarctica The Annual Review of Earth and Planetary Sciences is online at earth.annualreviews.org Abstract This article’s doi: Psychrophilic (cold-adapted) microorganisms make a major contribution 10.1146/annurev-earth-040610-133514 to Earth’s biomass and perform critical roles in global biogeochemical cy- Copyright c 2013 by Annual Reviews. cles. The vast extent and environmental diversity of Earth’s cold biosphere All rights reserved has selected for equally diverse microbial assemblages that can include ar- Access provided by University of Nevada - Reno on 05/25/15. For personal use only. Annu. Rev. Earth Planet. Sci. 2013.41:87-115. Downloaded from www.annualreviews.org chaea, bacteria, eucarya, and viruses. Underpinning the important ecological roles of psychrophiles are exquisite mechanisms of physiological adaptation. Evolution has also selected for cold-active traits at the level of molecular adaptation, and enzymes from psychrophiles are characterized by specific structural, functional, and stability properties. These characteristics of en- zymes from psychrophiles not only manifest in efficient low-temperature activity, but also result in a flexible protein structure that enables biocatalysis in nonaqueous solvents. -

Reporting Key Features in Cold-Adapted Bacteria
life Review Reporting Key Features in Cold-Adapted Bacteria Paula M. Tribelli 1,2 and Nancy I. López 1,2,* ID 1 Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, C1428EGA Buenos Aires, Argentina; [email protected] 2 IQUIBICEN, CONICET, C1428EGA Buenos Aires, Argentina * Correspondence: [email protected]; Tel.: +54-11-4576-3334 Received: 30 January 2018; Accepted: 12 March 2018; Published: 13 March 2018 Abstract: It is well known that cold environments are predominant over the Earth and there are a great number of reports analyzing bacterial adaptations to cold. Most of these works are focused on characteristics traditionally involved in cold adaptation, such as the structural adjustment of enzymes, maintenance of membrane fluidity, expression of cold shock proteins and presence of compatible solutes. Recent works based mainly on novel “omic” technologies have presented evidence of the presence of other important features to thrive in cold. In this work, we analyze cold-adapted bacteria, looking for strategies involving novel features, and/or activation of non-classical metabolisms for a cold lifestyle. Metabolic traits related to energy generation, compounds and mechanisms involved in stress resistance and cold adaptation, as well as characteristics of the cell envelope, are analyzed in heterotrophic cold-adapted bacteria. In addition, metagenomic, metatranscriptomic and metaproteomic data are used to detect key functions in bacterial communities inhabiting cold environments. Keywords: psychrophile; energy generation; polyhydroxyalkanoates; cell envelopes; cold-adapted bacterial communities 1. Introduction Temperature is a key factor for bacterial survival. Cold environments are predominant over the Earth. -

Draft Genome Sequence of Psychrobacter Sp. ENNN9 III, a Strain Isolated from Water in a Polluted Temperate Estuarine System (Ria De Aveiro, Portugal)
Genomics Data 8 (2016) 21–24 Contents lists available at ScienceDirect Genomics Data journal homepage: www.elsevier.com/locate/gdata Draft genome sequence of Psychrobacter sp. ENNN9_III, a strain isolated from water in a polluted temperate estuarine system (Ria de Aveiro, Portugal) Jaqueline Conceição Meireles Gomes a, Juliana Simão Nina de Azevedo b, Adonney Allan de Oliveira Veras a, Jorianne Thyeska Castro Alves a,IsabelHenriquesc, António Correia d, Artur Luiz da Costa da Silva a, Adriana Ribeiro Carneiro a,⁎ a Instituto de Ciências Biológicas, Universidade Federal do Pará (UFPA), Belém, Pará, Brazil b Departamento de Biologia, Universidade Federal Rural da Amazônia (UFRA), Capanema, Pará, Brazil c Departamento de Biologia, CESAM e iBiMED, Universidade de Aveiro, Aveiro, Portugal d Departamento de Biologia e CESAM, Universidade de Aveiro, Aveiro, Portugal article info abstract Article history: The genus Psychrobacter includes Gram-negative coccobacilli that are non-pigmented, oxidase-positive, non- Received 20 January 2016 motile, psychrophilic or psychrotolerant, and halotolerant. Psychrobacter strain ENNN9_III was isolated from Received in revised form 18 February 2016 water in a polluted temperate estuarine system, contaminated with hydrocarbons and heavy metals. The genome Accepted 20 February 2016 has a G + C content of 42.7%, 2618 open reading frames (ORFs), three copies of the rRNAs operon, and 29 tRNA Available online 23 February 2016 genes. Twenty-five sequences related to the degradation of aromatic compounds were predicted, as well as numerous Keywords: Psychrobacter genes related to resistance to metals or metal(loid)s. The genome sequence of Psychrobacter strain ENNN9_III Water provides the groundwork for further elucidation of the mechanisms of metal resistance and aromatic compounds Hydrocarbons degradation. -

Host-Microbe Interactions in Brine Shrimp Artemia Cultured Under Gnotobiotic and Conventional Rearing Conditions
Host-microbe interactions in brine shrimp Artemia cultured under gnotobiotic and conventional rearing conditions Niu Yufeng Thesis submitted in fulfillment of the requirements for the degree of Doctor (PhD) in Applied Biological Sciences: Environmental Technology Part of the work done during the PhD is being submitted as a patent application. Therefore, non UGent jury members will be asked to sign a confidential disclosure agreement, and all draft versions of the manuscript are to be treated as confidential material. Promoter: Prof. Dr. ir. Peter Bossier (Ghent University); Prof. Dr. ir. Tom Van de Wiele (Ghent University); Prof. Dr. Shuanglin Dong (Ocean University of China) Yufeng Niu. Host-microbe interactions in brine shrimp Artemia cultured under gnotobiotic and conventional rearing conditions. Dissertation, Ghent University, Belgium. Dutch translation of the title: Gastheer-microbe interacties in het pekelkreeftje Artemia in gnotobiotische en conventionele condities ISBN: 978-90-5989-709-0 The author and the promoters give the authorisation to consult and copy parts of this work for personal use only. Every other use is subject to the copyright laws. Permission to reproduce any material contained in this work should be obtained from the author. Yufeng Niu was funded by the Chinese Scholarship Council (CSC) and Special Research Fund (BOF) of Ghent University (BOF12/DC1/013; 01DI1312) TABLE OF CONTENTS LIST OF ABBREVIATIONS AND UNITS .................................................................................................. -

Free-Living Psychrophilic Bacteria of the Genus Psychrobacter Are
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.23.352302; this version posted October 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Title: Free-living psychrophilic bacteria of the genus Psychrobacter are 2 descendants of pathobionts 3 4 Running Title: psychrophilic bacteria descended from pathobionts 5 6 Daphne K. Welter1, Albane Ruaud1, Zachariah M. Henseler1, Hannah N. De Jong1, 7 Peter van Coeverden de Groot2, Johan Michaux3,4, Linda Gormezano5¥, Jillian L. Waters1, 8 Nicholas D. Youngblut1, Ruth E. Ley1* 9 10 1. Department of Microbiome Science, Max Planck Institute for Developmental 11 Biology, 12 Tübingen, Germany. 13 2. Department of Biology, Queen’s University, Kingston, Ontario, Canada. 14 3. Conservation Genetics Laboratory, University of Liège, Liège, Belgium. 15 4. Centre de Coopération Internationale en Recherche Agronomique pour le 16 Développement (CIRAD), UMR ASTRE, Montpellier, France. 17 5. Department of Vertebrate Zoology, American Museum of Natural History, New 18 York, NY, USA. 19 ¥deceased 20 *Correspondence: [email protected] 21 22 Abstract 23 Host-adapted microbiota are generally thought to have evolved from free-living 24 ancestors. This process is in principle reversible, but examples are few. The genus 25 Psychrobacter (family Moraxellaceae, phylum Gamma-Proteobacteria) includes species 26 inhabiting diverse and mostly polar environments, such as sea ice and marine animals. To 27 probe Psychrobacter’s evolutionary history, we analyzed 85 Psychrobacter strains by 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.23.352302; this version posted October 25, 2020. -

UNIVERSITY of NEVADA, RENO Dimensions of Antarctic Microbial
UNIVERSITY OF NEVADA, RENO Dimensions of Antarctic microbial life revealed through microscopic, cultivation-based, molecular phylogenetic and environmental genomic characterization A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biochemistry by Emanuele Kuhn Dr. Alison E. Murray/Dissertation Advisor May, 2014 Copyright by Emanuele Kuhn 2014 All Rights Reserved P age | i Abstract Extreme cold temperatures have shaped Antarctic environments and the life that lives within them. Microorganisms affiliated with the three domains of life - Bacteria, Archaea, and Eukaryote - can be found in Antarctic environments from deep subglacial lakes to dry deserts and from deep oceans to cold and dark winter surface seawaters. This dissertation focused on the investigation of the microbial assemblage in two Antarctic environments: Lake Vida, located in the McMurdo Dry Valleys, and the surface seawater from the Antarctic Peninsula. Lake Vida has a thick (27+ m) ice cover which seals a cryogenic brine reservoir within the lake ice below 16 m. This brine’s environment challenges the conditions for the existence of life. Despite the perceived challenges of aphotic, anoxic and freezing conditions, the brine contained an abundant assemblage (6.13 ± 1.65 × 10 7 cells mL -1) of ultra-small cells 0.192 ± 0.065 μm in diameter and a less abundant assemblage (1.47 ± 0.25 × 10 5 cells mL -1) of microbial cells ranging from > 0.2 to 1.5 μm in length. Scanning electron microscopy provided supporting evidence for cell membranes associated with the ~ 0.2 μm cells and helped discern a second smaller size class of particles (0.084 ± 0.063 μm). -

Marine Drugs
marine drugs Article Structure and In Vitro Bioactivity against Cancer Cells of the Capsular Polysaccharide from the Marine Bacterium Psychrobacter marincola Maxim S. Kokoulin 1,* , Alexandra S. Kuzmich 1, Lyudmila A. Romanenko 1, Irina V. Chikalovets 1,2 and Oleg V. Chernikov 1 1 G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch, Russian Academy of Sciences, 159/2, Prospect 100 let Vladivostoku, Vladivostok 690022, Russia; [email protected] (A.S.K.); [email protected] (L.A.R.); [email protected] (I.V.C.); [email protected] (O.V.C.) 2 Far Eastern Federal University, 8, Sukhanova str., Vladivostok 690950, Russia * Correspondence: [email protected]; Fax: +7-4232-314050 Received: 30 April 2020; Accepted: 18 May 2020; Published: 19 May 2020 Abstract: Psychrobacter marincola KMM 277T is a psychrophilic Gram-negative bacterium that has been isolated from the internal tissues of an ascidian Polysyncraton sp. Here, we report the structure of the capsular polysaccharide from P. marincola KMM 277T and its effect on the viability and colony formation of human acute promyelocytic leukemia HL-60 cells. The polymer was purified by several separation methods, including ultracentrifugation and chromatographic procedures, and the structure was elucidated by means of chemical analysis, 1-D, and 2-D NMR spectroscopy techniques. It was found that the polysaccharide consists of branched hexasaccharide repeating units containing two 2-N-acetyl-2-deoxy-d-galacturonic acids, and one of each of 2-N-acetyl-2-deoxy-d-glucose, d-glucose, d-ribose, and 7-N-acetylamino-3,5,7,9-tetradeoxy-5-N-[(R)-2-hydroxypropanoylamino]- l-glycero-l-manno-non-2-ulosonic acid.