Molecular Phylogenetic Analyses of Ecdysozoa and Haemosporida
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

First Description of Cryptosporidium Parvum in Carrier Pigeons
Veterinary Parasitology 243 (2017) 148–150 Contents lists available at ScienceDirect Veterinary Parasitology journal homepage: www.elsevier.com/locate/vetpar Research paper First description of Cryptosporidium parvum in carrier pigeons (Columba livia) MARK Bruno César Miranda Oliveiraa, Elis Domingos Ferraria, Mariele Fernanda da Cruz Panegossia, Alex Akira Nakamuraa, Flávio Sader Corbuccia, Walter Bertequini Nagataa, Bianca Martins dos Santosb, Jancarlo Ferreira Gomesb, Marcelo Vasconcelos Meirelesa, ⁎ Giovanni Widmerc, Katia Denise Saraiva Brescianid, a Universidade Estadual Paulista (Unesp), Faculdade de Medicina Veterinária, Araçatuba, Brazil b Laboratory of Visual Informatics in Biomedical and Health, Institute of Computing, University of Campinas – UNICAMP, São Paulo, Brazil c Department of Infectious Disease & Global Health, Cummings School of Veterinary Medicine at Tufts University, North Grafton, MA, USA d Universidade Estadual Paulista (Unesp), Faculdade de Medicina Veterinária, Araçatuba. Rua Clóvis Pestana, 793, Jardim Dona Amélia, cep 16050-680, Araçatuba, São Paulo, Brazil ARTICLE INFO ABSTRACT Keywords: The carrier pigeon and the domestic pigeon are different breeds of the species Columba livia. Carrier pigeons are Birds used for recreational activities such as bird contests and exhibitions. Due to the close contact with humans, these Carrier pigeons birds may potentially represent a public health risk, since they can host and disseminate zoonotic parasites, such Columba livia as those belonging to the genus Cryptosporidium (phylum Apicomplexa). The purpose of this work was the de- Cryptosporidiosis tection by microscopic and molecular techniques of Cryptosporidium spp. oocysts in fecal samples of carrier Cryptosporidium parvum pigeons, and subsequently to sequence the 18S ribosomal RNA marker of positive samples to identify the species. Nested PCR A total of 100 fecal samples were collected individually in two pigeon breeding facilities from Formiga and Araçatuba, cities located in Minas Gerais state and São Paulo state, Brazil, respectively. -

And Toxoplasmosis in Jackass Penguins in South Africa
IMMUNOLOGICAL SURVEY OF BABESIOSIS (BABESIA PEIRCEI) AND TOXOPLASMOSIS IN JACKASS PENGUINS IN SOUTH AFRICA GRACZYK T.K.', B1~OSSY J.].", SA DERS M.L. ', D UBEY J.P.···, PLOS A .. ••• & STOSKOPF M. K .. •••• Sununary : ReSlIlIle: E x-I1V\c n oN l~ lIrIUSATION D'Ar\'"TIGENE DE B ;IB£,'lA PH/Re El EN ELISA ET simoNi,cATIVlTli t'OUR 7 bxo l'l.ASMA GONIJfI DE SI'I-IENICUS was extracted from nucleated erythrocytes Babesia peircei of IJEMIiNSUS EN ArRIQUE D U SUD naturally infected Jackass penguin (Spheniscus demersus) from South Africo (SA). Babesia peircei glycoprotein·enriched fractions Babesia peircei a ele extra it d 'erythrocytes nue/fies p,ovenanl de Sphenicus demersus originoires d 'Afrique du Sud infectes were obto ined by conca navalin A-Sepharose affinity column natulellement. Des fractions de Babesia peircei enrichies en chromatogrophy and separated by sod ium dodecyl sulphate glycoproleines onl ele oblenues par chromatographie sur colonne polyacrylam ide gel electrophoresis (SDS-PAGE ). At least d 'alfinite concona valine A-Sephorose et separees par 14 protein bonds (9, 11, 13, 20, 22, 23, 24, 43, 62, 90, electrophorese en gel de polyacrylamide-dodecylsuJfale de sodium 120, 204, and 205 kDa) were observed, with the major protein (SOS'PAGE) Q uotorze bandes proleiques au minimum ont ete at 25 kDa. Blood samples of 191 adult S. demersus were tes ted observees (9, 1 I, 13, 20, 22, 23, 24, 43, 62, 90, 120, 204, by enzyme-linked immunosorbent assoy (ELISA) utilizing B. peircei et 205 Wa), 10 proleine ma;eure elant de 25 Wo. -

Diversity of Millipedes Along the Northern Western Ghats
Journal of Entomology and Zoology Studies 2014; 2 (4): 254-257 ISSN 2320-7078 Diversity of millipedes along the Northern JEZS 2014; 2 (4): 254-257 © 2014 JEZS Western Ghats, Rajgurunagar (MS), India Received: 14-07-2014 Accepted: 28-07-2014 (Arthropod: Diplopod) C. R. Choudhari C. R. Choudhari, Y.K. Dumbare and S.V. Theurkar Department of Zoology, Hutatma Rajguru Mahavidyalaya, ABSTRACT Rajgurunagar, University of Pune, The different vegetation type was used to identify the oligarchy among millipede species and establish India P.O. Box 410505 that millipedes in different vegetation types are dominated by limited set of species. In the present Y.K. Dumbare research elucidates the diversity of millipede rich in part of Northern Western Ghats of Rajgurunagar Department of Zoology, Hutatma (MS), India. A total four millipedes, Harpaphe haydeniana, Narceus americanus, Oxidus gracilis, Rajguru Mahavidyalaya, Trigoniulus corallines taxa belonging to order Polydesmida and Spirobolida; 4 families belongs to Rajgurunagar, University of Pune, Xystodesmidae, Spirobolidae, Paradoxosomatidae and Trigoniulidae and also of 4 genera were India P.O. Box 410505 recorded from the tropical or agricultural landscape of Northern Western Ghats. There was Harpaphe haydeniana correlated to the each species of millipede which were found in Northern Western Ghats S.V. Theurkar region of Rajgurunagar. At the time of diversity study, Trigoniulus corallines were observed more than Senior Research Fellowship, other millipede species, which supports the environmental determinism condition. Narceus americanus Department of Zoology, Hutatma was single time occurred in the agricultural vegetation landscape due to the geographical location and Rajguru Mahavidyalaya, habitat differences. Rajgurunagar, University of Pune, India Keywords: Diplopod, Northern Western Ghats, millipede diversity, Narceus americanus, Trigoniulus corallines 1. -

An Intestinal Gregarine of Nothria Conchylega (Polychaeta, Onuphidae)
Journal of Invertebrate Pathology 104 (2010) 172–179 Contents lists available at ScienceDirect Journal of Invertebrate Pathology journal homepage: www.elsevier.com/locate/jip Description of Trichotokara nothriae n. gen. et sp. (Apicomplexa, Lecudinidae) – An intestinal gregarine of Nothria conchylega (Polychaeta, Onuphidae) Sonja Rueckert *, Brian S. Leander Canadian Institute for Advanced Research, Program in Integrated Microbial Biodiversity, Departments of Botany and Zoology, University of British Columbia, #3529 – 6270 University Blvd., Vancouver, BC, Canada V6T 1Z4 article info abstract Article history: The trophozoites of a novel gregarine apicomplexan, Trichotokara nothriae n. gen. et sp., were isolated and Received 12 November 2009 characterized from the intestines of the onuphid tubeworm Nothria conchylega (Polychaeta), collected at Accepted 11 March 2010 20 m depth from the North-eastern Pacific Coast. The trophozoites were 50–155 lm long with a mid-cell Available online 23 March 2010 indentation that formed two prominent bulges (anterior bulge, 14–48 lm wide; posterior bulge, 15– 55 lm wide). Scanning electron microscopy (SEM) demonstrated that approximately 400 densely packed, Keywords: longitudinal epicytic folds (5 folds/lm) inscribe the surface of the trophozoites, and a prominently elon- Alveolata gated mucron (14–60 lm long and 6–12 lm wide) was covered with hair-like projections (mean length, Apicomplexa 1.97 m; mean width, 0.2 m at the base). Although a septum occurred at the junction between the cell Lecudinidae l l Lecudina proper and the mucron in most trophozoites, light microscopy (LM) demonstrated that the cell proper Parasite extended into the core of the mucron as a thin prolongation. -

Reconstruction of the Evolutionary History of Haemosporida
Parasitology International 65 (2016) 5–11 Contents lists available at ScienceDirect Parasitology International journal homepage: www.elsevier.com/locate/parint Reconstruction of the evolutionary history of Haemosporida (Apicomplexa) based on the cyt b gene with characterization of Haemocystidium in geckos (Squamata: Gekkota) from Oman João P. Maia a,b,c,⁎, D. James Harris a,b, Salvador Carranza c a CIBIO Research Centre in Biodiversity and Genetic Resources, InBIO, Universidade do Porto, Campus Agrário de Vairão, Rua Padre Armando Quintas, N° 7, 4485-661 Vairão, Vila do Conde, Portugal b Departamento de Biologia, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre FC4 4169-007 Porto, Portugal c Institut de Biologia Evolutiva (CSIC-Universitat Pompeu Fabra), Passeig Maritím de la Barceloneta, 37-49, 08003 Barcelona, Spain article info abstract Article history: The order Haemosporida (Apicomplexa) includes many medically important parasites. Knowledge on the diver- Received 4 April 2015 sity and distribution of Haemosporida has increased in recent years, but remains less known in reptiles and their Received in revised form 7 September 2015 taxonomy is still uncertain. Further, estimates of evolutionary relationships of this order tend to change when Accepted 10 September 2015 new genes, taxa, outgroups or alternative methodologies are used. We inferred an updated phylogeny for the Available online 12 September 2015 Cytochrome b gene (cyt b) of Haemosporida and screened a total of 80 blood smears from 17 lizard species from Oman belonging to 11 genera. The inclusion of previously underrepresented genera resulted in an alterna- Keywords: Haemoproteus tive estimate of phylogeny for Haemosporida based on the cyt b gene. -

Phylogeny of Molting Protostomes (Ecdysozoa) As Inferred from 18S and 28S Rrna Gene Sequences N
Molecular Biology, Vol. 39, No. 4, 2005, pp. 503–513. Translated from Molekulyarnaya Biologiya, Vol. 39, No. 4, 2005, pp. 590–601. Original Russian Text Copyright © 2005 by Petrov, Vladychenskaya. REVIEW AND EXPERIMANTAL ARTICLES UDC 575.852'113;595.131'132'145.2'185;595.2 Phylogeny of Molting Protostomes (Ecdysozoa) as Inferred from 18S and 28S rRNA Gene Sequences N. B. Petrov and N. S. Vladychenskaya Belozersky Institute of Physicochemical Biology, Moscow State University, Moscow, 119992 Russia e-mail: [email protected] Received December 29, 2004 Abstract—Phylogenetic relationships within the group of molting protostomes were reconstructed by compar- ing the sets of 18S and 28S rRNA gene sequences considered either separately or in combination. The reliability of reconstructions was estimated from the bootstrap indices for major phylogenetic tree nodes and from the degree of congruence of phylogenetic trees obtained by different methods. By either criterion, the phylogenetic trees reconstructed on the basis of both 18 and 28S rRNA gene sequences were better than those based on the 18S or 28S sequences alone. The results of reconstruction are consistent with the phylogenetic hypothesis clas- sifying protostomes into two major clades: molting Ecdysozoa (Priapulida + Kinorhyncha, Nematoda + Nema- tomorpha, Onychophora + Tardigrada, Myriapoda + Chelicerata, and Crustacea + Hexapoda) and nonmolting Lophotrochozoa (Plathelminthes, Nemertini, Annelida, Mollusca, Echiura, and Sipuncula). Nematomorphs (Nematomorpha) do not belong to the clade Cephalorhyncha (Priapulida + Kinorhyncha). It is concluded that combined data on the 18S and 28S rRNA gene sequences provide a more reliable basis for phylogenetic infer- ences. Key words: 18S rRNA, 28S rRNA, molecular phylogeny, Protostomia, Ecdysozoa, Arthropoda, Cephalorhyn- cha, Nematoda, Nematomorpha INTRODUCTION accepted in zoology and became basic in textbooks Since its origin at the turn of the 20th century, [12, 13]. -

Feline Immune Response to Infection with Cytauxzoon Felis and The
FELINE IMMUNE RESPONSE TO INFECTION WITH CYTAUXZOON FELIS AND THE ROLE OF CD18 IN THE PATHOGENESIS OF CYTAUXZOONOSIS by KARELMA FRONTERA-ACEVEDO (Under the Direction of Kaori Sakamoto) ABSTRACT Cytauxzoonosis is a highly fatal, hemoprotozoal disease of cats in the Mid-Western, Mid- Atlantic, and Southeastern United States, caused by Cytauxzoon felis. Although the causative agent has been recognized since 1976, no study has profiled the immune response of infected cats, there is no definitive cure, and C. felis has not been successfully maintained in cell cultures in vitro, thwarting research efforts. One of the main histopathologic characteristics of this disease is the presence of giant, infected, intravascular macrophages, many of which are adhered to the vascular endothelium. The main goals of this project are: 1) to characterize the feline immune response to C. felis; 2) to develop a cell culture system in order to study C. felis in vitro; and 3) to determine whether CD18 plays a role in the pathogenesis of cytauxzoonosis. INDEX WORDS: Cat, Cytauxzoon felis, pathogenesis, protozoal disease, veterinary pathology FELINE IMMUNE RESPONSE TO INFECTION WITH CYTAUXZOON FELIS AND THE ROLE OF CD18 IN THE PATHOGENESIS OF CYTAUXZOONOSIS by KARELMA FRONTERA-ACEVEDO BS, University of Florida, 2004 DVM, Louisiana State University, 2008 A Dissertation Submitted to the Graduate Faculty of The University of Georgia in Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY ATHENS, GEORGIA 2013 © 2013 Karelma Frontera-Acevedo All -
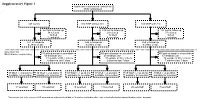
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines. -

The Nuclear 18S Ribosomal Dnas of Avian Haemosporidian Parasites Josef Harl1, Tanja Himmel1, Gediminas Valkiūnas2 and Herbert Weissenböck1*
Harl et al. Malar J (2019) 18:305 https://doi.org/10.1186/s12936-019-2940-6 Malaria Journal RESEARCH Open Access The nuclear 18S ribosomal DNAs of avian haemosporidian parasites Josef Harl1, Tanja Himmel1, Gediminas Valkiūnas2 and Herbert Weissenböck1* Abstract Background: Plasmodium species feature only four to eight nuclear ribosomal units on diferent chromosomes, which are assumed to evolve independently according to a birth-and-death model, in which new variants origi- nate by duplication and others are deleted throughout time. Moreover, distinct ribosomal units were shown to be expressed during diferent developmental stages in the vertebrate and mosquito hosts. Here, the 18S rDNA sequences of 32 species of avian haemosporidian parasites are reported and compared to those of simian and rodent Plasmodium species. Methods: Almost the entire 18S rDNAs of avian haemosporidians belonging to the genera Plasmodium (7), Haemo- proteus (9), and Leucocytozoon (16) were obtained by PCR, molecular cloning, and sequencing ten clones each. Phy- logenetic trees were calculated and sequence patterns were analysed and compared to those of simian and rodent malaria species. A section of the mitochondrial CytB was also sequenced. Results: Sequence patterns in most avian Plasmodium species were similar to those in the mammalian parasites with most species featuring two distinct 18S rDNA sequence clusters. Distinct 18S variants were also found in Haemopro- teus tartakovskyi and the three Leucocytozoon species, whereas the other species featured sets of similar haplotypes. The 18S rDNA GC-contents of the Leucocytozoon toddi complex and the subgenus Parahaemoproteus were extremely high with 49.3% and 44.9%, respectively. -

Why the –Omic Future of Apicomplexa Should Include Gregarines Julie Boisard, Isabelle Florent
Why the –omic future of Apicomplexa should include Gregarines Julie Boisard, Isabelle Florent To cite this version: Julie Boisard, Isabelle Florent. Why the –omic future of Apicomplexa should include Gregarines. Biology of the Cell, Wiley, 2020, 10.1111/boc.202000006. hal-02553206 HAL Id: hal-02553206 https://hal.archives-ouvertes.fr/hal-02553206 Submitted on 24 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article title: Why the –omic future of Apicomplexa should include Gregarines. Names of authors: Julie BOISARD1,2 and Isabelle FLORENT1 Authors affiliations: 1. Molécules de Communication et Adaptation des Microorganismes (MCAM, UMR 7245), Département Adaptations du Vivant (AVIV), Muséum National d’Histoire Naturelle, CNRS, CP52, 57 rue Cuvier 75231 Paris Cedex 05, France. 2. Structure et instabilité des génomes (STRING UMR 7196 CNRS / INSERM U1154), Département Adaptations du vivant (AVIV), Muséum National d'Histoire Naturelle, CP 26, 57 rue Cuvier 75231 Paris Cedex 05, France. Short Title: Gregarines –omics for Apicomplexa studies -

Late Eocene Potamogalidae and Tenrecidae (Mammalia) from the Sperrgebiet, Namibia
Late Eocene Potamogalidae and Tenrecidae (Mammalia) from the Sperrgebiet, Namibia Martin Pickford Sorbonne Universités (CR2P, UMR 7207 du CNRS, Département Histoire de la Terre, Muséum National d’Histoire Naturelle et Université Pierre et Marie Curie) case postale 38, 57 rue Cuvier, 75005 Paris. e-mail: < [email protected] > Abstract : The Late Eocene (Bartonian) Eocliff Limestone has yielded a rich, diverse and well- preserved micromammalian fauna which includes three tenrecoids, a chrysochlorid, several macroscelidids and at least eight taxa of rodents. The available cranio-dental and post-cranial elements reveal that the three tenrecoid species are closely related to potamogalids (one taxon) and to tenrecids (two taxa). The dichotomy between these two families probably occurred a long time before deposition of the Eocliff carbonate, possibly during the Palaeocene or even as early as the Late Cretaceous. The dentitions of the Eocliff potamogalid and tenrecids exhibit primitive versions of protozalambdodonty, in which the upper molars have clear metacones. Three new genera and species are described. Key Words : Potamogalidae, Tenrecidae, Zalambdodonty, Late Eocene, Namibia, Evolution To cite this paper: Pickford, M., 2015. Late Eocene Potamogalidae and Tenrecidae (Mammalia) from the Sperrgebiet, Namibia. Co mmunications of the Geological Survey of Namibia , 16, 114-152. Submitted in 2015. Introduction the suborder Tenrecoidea is not well represented in North Africa. The Late Eocene The discovery of Bartonian vertebrates Namibian fossils thus help to fill extensive in the Sperrgebiet, Namibia, is of major chronological and geographic gaps in the significance for throwing light on the evolution history and distribution of zalambdodont of African Palaeogene mammals, especially mammals in Africa, although the geographic that of rodents, tenrecoids and chrysochlorids position of the deposits from which they were (Pickford et al. -

Subterranean Mammals Show Convergent Regression in Ocular Genes and Enhancers, Along with Adaptation to Tunneling
RESEARCH ARTICLE Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling Raghavendran Partha1, Bharesh K Chauhan2,3, Zelia Ferreira1, Joseph D Robinson4, Kira Lathrop2,3, Ken K Nischal2,3, Maria Chikina1*, Nathan L Clark1* 1Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, United States; 2UPMC Eye Center, Children’s Hospital of Pittsburgh, Pittsburgh, United States; 3Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, United States; 4Department of Molecular and Cell Biology, University of California, Berkeley, United States Abstract The underground environment imposes unique demands on life that have led subterranean species to evolve specialized traits, many of which evolved convergently. We studied convergence in evolutionary rate in subterranean mammals in order to associate phenotypic evolution with specific genetic regions. We identified a strong excess of vision- and skin-related genes that changed at accelerated rates in the subterranean environment due to relaxed constraint and adaptive evolution. We also demonstrate that ocular-specific transcriptional enhancers were convergently accelerated, whereas enhancers active outside the eye were not. Furthermore, several uncharacterized genes and regulatory sequences demonstrated convergence and thus constitute novel candidate sequences for congenital ocular disorders. The strong evidence of convergence in these species indicates that evolution in this environment is recurrent and predictable and can be used to gain insights into phenotype–genotype relationships. DOI: https://doi.org/10.7554/eLife.25884.001 *For correspondence: [email protected] (MC); [email protected] (NLC) Competing interests: The Introduction authors declare that no The subterranean habitat has been colonized by numerous animal species for its shelter and unique competing interests exist.