The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fast Pressure-Jump All-Atom Simulations and Experiments Reveal Site-Specific Protein Dehydration-Folding Dynamics
Fast pressure-jump all-atom simulations and experiments reveal site-specific protein dehydration-folding dynamics Maxim B. Prigozhina,1,2, Yi Zhangb,1, Klaus Schultenb,c, Martin Gruebelea,b,c,3, and Taras V. Pogorelova,b,d,e,3 aDepartment of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801; bBeckman Institute for Advanced Science and Technology, Center for Biophysics and Quantitative Biology, University of Illinois at Urbana–Champaign, Urbana, IL 61801; cDepartment of Physics, University of Illinois at Urbana–Champaign, Urbana, IL 61801; dNational Center for Supercomputing Applications, University of Illinois at Urbana–Champaign, Urbana, IL 61801; and eSchool of Chemical Sciences, University of Illinois at Urbana–Champaign, Urbana, IL 61801 Edited by José N. Onuchic, Rice University, Houston, TX, and approved February 8, 2019 (received for review August 30, 2018) As theory and experiment have shown, protein dehydration is a and simulated using MD (18). Desolvated cavities in the protein major contributor to protein folding. Dehydration upon folding core (15) are restored by downward pressure jumps, which fa- can be characterized directly by all-atom simulations of fast cilitate refolding without a change in solvent or thermal motion. pressure drops, which create desolvated pockets inside the In addition, pressure perturbation reduces the effects of protein nascent hydrophobic core. Here, we study pressure-drop refolding aggregation, common in temperature-jump (T-jump) experi- of three λ-repressor fragment (λ6–85) mutants computationally and ments performed near the denaturation temperature (16, 17). experimentally. The three mutants report on tertiary structure for- Finally, pressure-jump (P-jump) experiments can utilize intrinsic mation via different fluorescent helix–helix contact pairs. -

Studying Backbone Torsional Dynamics of Intrinsically Disordered Proteins Using fluorescence Depolarization Kinetics
J Biosci Vol. 43, No. 3, July 2018, pp. 455–462 Ó Indian Academy of Sciences DOI: 10.1007/s12038-018-9766-1 Studying backbone torsional dynamics of intrinsically disordered proteins using fluorescence depolarization kinetics 1,3 1,2,3 DEBAPRIYA DAS and SAMRAT MUKHOPADHYAY * 1Centre for Protein Science, Design and Engineering, Indian Institute of Science Education and Research (IISER) Mohali, Punjab, India 2Department of Biological Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Punjab, India 3Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Punjab, India *Corresponding author (Email, [email protected]) Published online: 7 June 2018 Intrinsically disordered proteins (IDPs) do not autonomously adopt a stable unique 3D structure and exist as an ensemble of rapidly interconverting structures. They are characterized by significant conformational plasticity and are associated with several biological functions and dysfunctions. The rapid conformational fluctuation is governed by the backbone segmental dynamics arising due to the dihedral angle fluctuation on the Ramachandran /–w conformational space. We discovered that the intrinsic backbone torsional mobility can be monitored by a sensitive fluorescence readout, namely fluorescence depolarization kinetics, of tryptophan in an archetypal IDP such as a-synuclein. This methodology allows us to map the site-specific torsional mobility in the dihedral space within picosecond-nanosecond time range at a low protein concen- tration under the native condition. The characteristic timescale of * 1.4 ns, independent of residue position, represents collective torsional dynamics of dihedral angles (/ and w) of several residues from tryptophan and is independent of overall global tumbling of the protein. -

Linking Predictions of Protein Structure and Disorder Through Molecular Simulation
The Order-Disorder Continuum: Linking Predictions of Protein Structure and Disorder through Molecular Simulation The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Hsu, Claire C., Markus J. Buehler, and Anna Tarakanova. "The Order-Disorder Continuum: Linking Predictions of Protein Structure and Disorder through Molecular Simulation." Scientific Reports, 10 (February 2020): 2068. © 2020, The Author(s). As Published http://dx.doi.org/10.1038/s41598-020-58868-w Publisher Springer Science and Business Media LLC Citable link https://hdl.handle.net/1721.1/125166 Terms of Use Creative Commons Attribution 4.0 International license Detailed Terms https://creativecommons.org/licenses/by/4.0/ www.nature.com/scientificreports OPEN The Order-Disorder Continuum: Linking Predictions of Protein Structure and Disorder through Molecular Simulation Claire C. Hsu1, Markus J. Buehler2 & Anna Tarakanova3,4* Intrinsically disordered proteins (IDPs) and intrinsically disordered regions within proteins (IDRs) serve an increasingly expansive list of biological functions, including regulation of transcription and translation, protein phosphorylation, cellular signal transduction, as well as mechanical roles. The strong link between protein function and disorder motivates a deeper fundamental characterization of IDPs and IDRs for discovering new functions and relevant mechanisms. We review recent advances in experimental techniques that have improved identifcation of disordered regions -

The Conformation of Serum Albumin in Solution: a Combined Phosphorescence Depolarization-Hydrodynamic Modeling Study
2422 Biophysical Journal Volume 80 May 2001 2422–2430 The Conformation of Serum Albumin in Solution: A Combined Phosphorescence Depolarization-Hydrodynamic Modeling Study M. Luisa Ferrer,* Ricardo Duchowicz,* Beatriz Carrasco,† Jose´ Garcı´a de la Torre,† and A. Ulises Acun˜a* *Instituto Quı´mica-Fı´sica “Rocasolano”, Consejo Superior de Investigaciones Cientificas, 28006 Madrid, and †Departamento de Quı´mica-Fı´sica, Universidad de Murcia, 30071 Murcia, Spain ABSTRACT There is a striking disparity between the heart-shaped structure of human serum albumin (HSA) observed in single crystals and the elongated ellipsoid model used for decades to interpret the protein solution hydrodynamics at neutral pH. These two contrasting views could be reconciled if the protein were flexible enough to change its conformation in solution from that found in the crystal. To investigate this possibility we recorded the rotational motions in real time of an erythrosin- bovine serum albumin complex (Er-BSA) over an extended time range, using phosphorescence depolarization techniques. These measurements are consistent with the absence of independent motions of large protein segments in solution, in the time range from nanoseconds to fractions of milliseconds, and give a single rotational correlation time (BSA, 1 cP, 20°C) ϭ 40 Ϯ 2 ns. In addition, we report a detailed analysis of the protein hydrodynamics based on two bead-modeling methods. In the first, BSA was modeled as a triangular prismatic shell with optimized dimensions of 84 ϫ 84 ϫ 84 ϫ 31.5 Å, whereas in the second, the atomic-level structure of HSA obtained from crystallographic data was used to build a much more refined rough-shell model. -

Numerical Evaluation of Protein Global Vibrations at Terahertz Frequencies by Means of Elastic Lattice Models †
Proceedings Numerical Evaluation of Protein Global Vibrations at Terahertz Frequencies by Means of Elastic † Lattice Models Domenico Scaramozzino *, Giuseppe Lacidogna, Gianfranco Piana and Alberto Carpinteri Department of Structural, Geotechnical and Building Engineering, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy; [email protected] (G.L.); [email protected] (G.P.); [email protected] (A.C.) * Correspondence: [email protected] † Presented at 1st International Electronic Conference on Applied Sciences, 10–30 November 2020; Available online: https://asec2020.sciforum.net/. Published: 9 November 2020 Abstract: Proteins represent one of the most important building blocks for most biological processes. Their biological mechanisms have been found to correlate significantly with their dynamics, which is commonly investigated through molecular dynamics (MD) simulations. However, important insights on protein dynamics and biological mechanisms have also been obtained via much simpler and computationally efficient calculations based on elastic lattice models (ELMs). The application of structural mechanics approaches, such as modal analysis, to the protein ELMs has allowed to find impressive results in terms of protein dynamics and vibrations. The low-frequency vibrations extracted from the protein ELM are usually found to occur within the terahertz (THz) frequency range and correlate fairly accurately with the observed functional motions. In this contribution, the global vibrations of lysozyme will be investigated by means of a finite element (FE) truss model, and we will show that there exists complete consistency between the proposed FE approach and one of the more well-known ELMs for protein dynamics, the anisotropic network model (ANM). The proposed truss model can consequently be seen as a simple method, easily accessible to the structural mechanics community members, to analyze protein vibrations and their connections with the biological activity. -

Jacquelyn S. Fetrow
Jacquelyn S. Fetrow President and Professor of Chemistry Albright College Curriculum Vitae Office of the President Work Email: [email protected] Library and Administration Building Office Phone: 610-921-7600 N. 13th and Bern Streets, P.O. Box 15234 Reading, PA 19612 Education Ph.D. Biological Chemistry, December, 1986 B.S. Biochemistry, May, 1982 Department of Biological Chemistry Albright College, Reading, PA The Pennsylvania State University College of Medicine, Hershey, PA Graduated summa cum laude Loops: A Novel Class of Protein Secondary Structure Thesis Advisor: George D. Rose Professional Experience Albright College, Reading PA President and Professor of Chemistry June 2017-present University of Richmond, Richmond, VA Provost and Vice President of Academic Affairs July 2014-December 2016 Professor of Chemistry July 2014-May 2017 Responsibilities as Provost: Chief academic administrator for all academic matters for the University of Richmond, a university with five schools (Arts and Sciences, Business, Law, Leadership, and Professional and Continuing Studies), ~400 faculty and ~3300 full-time undergraduate and graduate students; manage the ~$91.8M annual operating budget of the Academic Affairs Division, as well as endowment and gift accounts; oversee Richmond’s Bonner Center of Civic Engagement (engage.richmond.edu), Center for International Education (international.richmond.edu), Registrar (registrar.richmond.edu), Office of Institutional Effectiveness (ifx.richmond.edu), as well as other programs and staff; partner with VP -

Dynamics of Proteins in Solution Biophysics
Quarterly Reviews of Dynamics of proteins in solution Biophysics Marco Grimaldo1,2, Felix Roosen-Runge1,3, Fajun Zhang2, Frank Schreiber2 cambridge.org/qrb and Tilo Seydel1 1Institut Max von Laue - Paul Langevin, 71 avenue des Martyrs, 38042 Grenoble, France; 2Institut für Angewandte Physik, Universität Tübingen, Auf der Morgenstelle 10, 72076 Tübingen, Germany and 3Division for Physical Review Chemistry, Lund University, Naturvetarvägen 14, 22100 Lund, Sweden Cite this article: Grimaldo M, Roosen-Runge F, Abstract Zhang F, Schreiber F, Seydel T (2019). Dynamics of proteins in solution. Quarterly The dynamics of proteins in solution includes a variety of processes, such as backbone and Reviews of Biophysics 52, e7, 1–63. https:// side-chain fluctuations, interdomain motions, as well as global rotational and translational doi.org/10.1017/S0033583519000027 (i.e. center of mass) diffusion. Since protein dynamics is related to protein function and essen- tial transport processes, a detailed mechanistic understanding and monitoring of protein Received: 15 February 2019 Revised: 27 February 2019 dynamics in solution is highly desirable. The hierarchical character of protein dynamics Accepted: 4 March 2019 requires experimental tools addressing a broad range of time- and length scales. We discuss how different techniques contribute to a comprehensive picture of protein dynamics, and Key words: focus in particular on results from neutron spectroscopy. We outline the underlying principles Quasielastic neutron spectroscopy; protein diffusion; protein -

Comparisons of Protein Dynamics from Experimental Structure
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital Repository @ Iowa State University Biochemistry, Biophysics and Molecular Biology Biochemistry, Biophysics and Molecular Biology Publications 1-27-2018 Comparisons of Protein Dynamics from Experimental Structure Ensembles, Molecular Dynamics Ensembles, and Coarse-Grained Elastic Network Models Kannan Sankar Iowa State University Sambit K. Mishra Iowa State University Robert L. Jernigan Iowa State University, [email protected] Follow this and additional works at: https://lib.dr.iastate.edu/bbmb_ag_pubs Part of the Biochemistry Commons, Bioinformatics Commons, Biophysics Commons, Molecular Biology Commons, and the Structural Biology Commons The ompc lete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ bbmb_ag_pubs/197. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Biochemistry, Biophysics and Molecular Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Biochemistry, Biophysics and Molecular Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Comparisons of Protein Dynamics from Experimental Structure Ensembles, Molecular Dynamics Ensembles, and Coarse-Grained Elastic Network Models Abstract Predicting protein motions is important for bridging the gap between protein structure and function. With growing numbers of structures of the same, or closely related proteins becoming available, it is now possible to understand more about the intrinsic dynamics of a protein with principal component analysis (PCA) of the motions apparent within ensembles of experimental structures. -
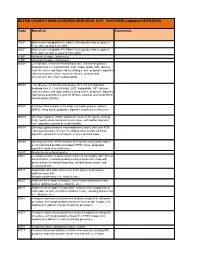
SETON CHARITY NON-COVERED SERVICES EFF: 01/01/2020 (Updated 05/05/2020)
SETON CHARITY NON-COVERED SERVICES EFF: 01/01/2020 (updated 05/05/2020) Code Narrative Comments 3051F Most recent hemoglobin A1c (HbA1c) level greater than or equal to 7.0% and less than 8.0% (DM) 3052F Most recent hemoglobin A1c (HbA1c) level greater than or equal to 8.0% and less than or equal to 9.0% (DM) 4185F Histamine therapy - intravenous 4186F Histamine therapy - intravenous 0002M Liver disease, ten biochemical assays (ALT, A2-macro- globulin, apolipoprotein A-1, total bilirubin, GGT, hapto- globin, AST, glucose, total cholesterol and triglycerides) utilizing serum, prognostic algorithm reported as quanti- tative scores for fibrosis, steatosis and alcoholic/non alco- holic steatohepatitis 0003M Liver disease, ten biochemical assays (ALT, A2-macroglobulin, apolipoprotein A-1, total bilirubin, GGT, haptoglobin, AST, glucose, total cholesterol and triglycerides) utilizing serum, prognostic algorithm reported as quantitative scores for fibrosis, steatosis and nonalcoholic steatohepatitis (NASH) 0004M Scoliosis, DNA analysis of 53 single nucleotide polymor- phisms (SNPs), using saliva, prognostic algorithm reported as a risk score 0006M Oncology (hepatic), mRNA expression levels of 161 genes, utilizing fresh hepatocellular carcinoma tumor tissue, with alpha-fetoprotein level, algorithm reported as a risk classifier 0007M Oncology (gastrointestinal neuroendocrine tumors), real- time PCR expression analysis of 51 genes, utilizing whole peripheral blood, algorithm reported as a nomogram of tumor disease index 0008M Oncology (breast), mRNA -

Nucleolar Proteome Dynamics Analysis
letters to nature GLT1 activity and immunoblotting against Borrelia spp. in the mouse brain and other sites. Antimicrob. Agents Chemother. 38, 2632–2636 Levels of GLT1 protein were quantified by immunoblots3. Functional glutamate transport (1996). was measured by accumulation of 3H-glutamate in spinal cord slice or crude cortical 15. Chen, W. et al. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures 25 and in neurons and astrocytes in the rat brain. J. Neurosci. 22, 2142–2152 (2002). synaptosomal membranes . Measurement of total glutamate uptake actually reflects the þ combined physiological activity of all transporter subtypes. GLT1 protein is uniquely 16. Schlag, B. D. et al. Regulation of the glial Na -dependent glutamate transporters by cyclic AMP sensitive to transport inhibition by dihydrokainate (DHK). To estimate the contribution analogs and neurons. Mol. Pharmacol. 53, 355–369 (1998). of GLT1 to transport, aliquots of tissue homogenates were also incubated with 300 mM 17. Guo, H. et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity DHK. Non-specific uptake was determined in the presence of 300 mM threo-b- and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 12, 2519–2532 (2003). 18. Romera, C. et al. In vitro ischemic tolerance involves upregulation of glutamate transport partly hydroxyaspartate (THA), at 0 8C and in sodium-free homogenates. mediated by the TACE/ADAM17-tumor necrosis factor-alpha pathway. J. Neurosci. 24, 1350–1357 Generation of GLT1 BAC eGFP transgenic mouse (2004). 19. Spalloni, A. et al. Cu/Zn-superoxide dismutase (GLY93 ! ALA) mutation alters AMPA receptor 26 The BAC transgenic mice were generated as described previously with a shuttle vector subunit expression and function and potentiates kainate-mediated toxicity in motor neurons in provided by N. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

The Development of the Prediction of Protein Structure
6 The Development of the Prediction of Protein Structure Gerald D. Fasman I. Introduction .................................................................... 194 II. Protein Topology. .. 196 III. Techniques of Protein Prediction ................................................... 198 A. Sequence Alignment .......................................................... 199 B. Hydrophobicity .............................................................. 200 C. Minimum Energy Calculations ................................................. 202 IV. Approaches to Protein Conformation ................................................ 203 A. Solvent Accessibility ......................................................... 203 B. Packing of Residues .......................................................... 204 C. Distance Geometry ........................................................... 205 D. Amino Acid Physicochemical Properties ......................................... 205 V. Prediction of the Secondary Structure of Proteins: a Helix, ~ Strands, and ~ Turn .......... 208 A. ~ Turns .................................................................... 209 B. Evaluation of Predictive Methodologies .......................................... 218 C. Other Predictive Algorithms ................................................... 222 D. Chou-Fasman Algorithm ...................................................... 224 E. Class Prediction ............................................................. 233 VI. Prediction of Tertiary Structure ...................................................