Ischemia”—Stressed PC12 Pheochromocytoma Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
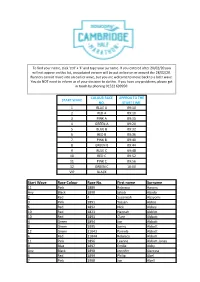
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Family Group Sheets Surname Index
PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS SURNAME INDEX This collection of 660 folders contains over 50,000 family group sheets of families that resided in Passaic and Bergen Counties. These sheets were prepared by volunteers using the Societies various collections of church, ceme tery and bible records as well as city directo ries, county history books, newspaper abstracts and the Mattie Bowman manuscript collection. Example of a typical Family Group Sheet from the collection. PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS — SURNAME INDEX A Aldous Anderson Arndt Aartse Aldrich Anderton Arnot Abbott Alenson Andolina Aronsohn Abeel Alesbrook Andreasen Arquhart Abel Alesso Andrews Arrayo Aber Alexander Andriesse (see Anderson) Arrowsmith Abers Alexandra Andruss Arthur Abildgaard Alfano Angell Arthurs Abraham Alje (see Alyea) Anger Aruesman Abrams Aljea (see Alyea) Angland Asbell Abrash Alji (see Alyea) Angle Ash Ack Allabough Anglehart Ashbee Acker Allee Anglin Ashbey Ackerman Allen Angotti Ashe Ackerson Allenan Angus Ashfield Ackert Aller Annan Ashley Acton Allerman Anners Ashman Adair Allibone Anness Ashton Adams Alliegro Annin Ashworth Adamson Allington Anson Asper Adcroft Alliot Anthony Aspinwall Addy Allison Anton Astin Adelman Allman Antoniou Astley Adolf Allmen Apel Astwood Adrian Allyton Appel Atchison Aesben Almgren Apple Ateroft Agar Almond Applebee Atha Ager Alois Applegate Atherly Agnew Alpart Appleton Atherson Ahnert Alper Apsley Atherton Aiken Alsheimer Arbuthnot Atkins Aikman Alterman Archbold Atkinson Aimone -

Localized Holes and Delocalized Electrons in Photoexcited Inorganic Perovskites: Watching Each Atomic Actor by Picosecond X-Ray Absorption Spectroscopy Fabio G
Localized holes and delocalized electrons in photoexcited inorganic perovskites: Watching each atomic actor by picosecond X-ray absorption spectroscopy Fabio G. Santomauro, Jakob Grilj, Lars Mewes, Georgian Nedelcu, Sergii Yakunin, Thomas Rossi, Gloria Capano, André Al Haddad, James Budarz, Dominik Kinschel, Dario S. Ferreira, Giacomo Rossi, Mario Gutierrez Tovar, Daniel Grolimund, Valerie Samson, Maarten Nachtegaal, Grigory Smolentsev, Maksym V. Kovalenko, and Majed Chergui Citation: Structural Dynamics 4, 044002 (2017); doi: 10.1063/1.4971999 View online: http://dx.doi.org/10.1063/1.4971999 View Table of Contents: http://aca.scitation.org/toc/sdy/4/4 Published by the American Institute of Physics Articles you may be interested in Structural enzymology using X-ray free electron lasers Structural Dynamics 4, 044003 (2016); 10.1063/1.4972069 A comparison of the innate flexibilities of six chains in F1-ATPase with identical secondary and tertiary folds; 3 active enzymes and 3 structural proteins Structural Dynamics 4, 044001 (2016); 10.1063/1.4967226 The image of scientists in The Big Bang Theory Physics Today 70, 40 (2017); 10.1063/PT.3.3427 Ultrafast electron crystallography of the cooperative reaction path in vanadium dioxide Structural Dynamics 3, 034304 (2016); 10.1063/1.4953370 A general method for baseline-removal in ultrafast electron powder diffraction data using the dual-tree complex wavelet transform Structural Dynamics 4, 044004 (2016); 10.1063/1.4972518 A beam branching method for timing and spectral characterization of hard X-ray free-electron lasers Structural Dynamics 3, 034301 (2016); 10.1063/1.4939655 STRUCTURAL DYNAMICS 4, 044002 (2017) Localized holes and delocalized electrons in photoexcited inorganic perovskites: Watching each atomic actor by picosecond X-ray absorption spectroscopy Fabio G. -
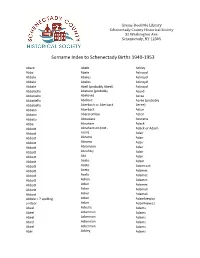
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -

Protocolo De Ensayo Clínico Con Medicamentos
Protocol PROFTMDECOL Study CLINICAL RESEARCH PROTOCOL Open-label, randomized study to assess the efficacy of a probiotic or fecal microbiota transplantation (FMT) on the eradication of rectal multidrug-resistant Gram-negative bacilli (MDR-GNB) carriage Protocol: PROFTMDECOL Version: 1.0 (02/12/2019) Sponsor: Fundació Clínic per a la Recerca Biomèdica Principal Investigator: Dra. Ana Del Río. Co-principal Investigator: Dr. José Antonio Martínez. Infectious Diseases Hospital Clínic Villarroel, 170 08036 Barcelona Version 1.0 dated 02 December 2019 - 1 - Protocol PROFTMDECOL Study 1. Brief description 1.0. Study This is an investigator-initiated, single center, parallel-group, randomized, open-label, clinical research with non pharmacological products. 1.1. Sponsor Fundació Clínic per a la Recerca Biomédica. Rosselló, 149 08036 Barcelona 1.2. Study identification Title: Open-label, randomized study to assess the efficacy of a probiotic or fecal microbiota transplantation (FMT) on the eradication of rectal multidrug-resistant Gram-negative bacilli (MDR-GNB) carriage. Protocol code: PROFTMDECOL 1.4. Investigators Principal investigator: Dra. Ana Del Río, Infectious Diseases Co-principal investigator: Dr. José Antonio Martínez, Infectious Diseases Collaborators: Dr. C. Pitart, Microbiología Clínica Dr. J. Mercadal, Anestesiología Dra. E. López, Farmacia Hospitalaria 1.5. Clinical sites Single centre study: Hospital Clínic, Barcelona 1.6. Ethics approval The protocol, informed consent form, and other required documents will be approved by the Ethics Committee (Comité de Ética de la Investigación, CEI) of the Hospital Clínic de Barcelona before enrolment of subjects in the study. 1.7. Monitoring Dr. JA Arnaiz, Unidad de Ensayos Clínicos (CTU Clinic) Farmacología Clínica, Hospital Clínic, Barcelona. -

Peer-Reviewed Articles______Works Are Reported in Chronological Order
Prof. Giulia Fulvia Mancini – List of Publications Peer-reviewed Articles_____________________________________________________________ Works are reported in chronological order. O. Cannelli, N. Colonna, M. Puppin, T. Rossi, D. Kinschel, L. Leroy, J. Löffler, A. M. March, G. Doumy, A. Al Haddad, M.-F. Tu, Y. Kumagai, D. Walko, G. Smolentsev, F. Krieg, S. C. Boehme, M. V. Kovalenko, M. Chergui, G. F. Mancini, "Quantifying Photoinduced Polaronic Distortions in Inorganic Lead Halide Perovskites Nanocrystals", J.Am.Chem. Soc. (2021) - Accepted. J. R. Rouxel, D. Fainozzi, R. Mankowsky, B. Rösner, G. Seniutinas, R. Mincigrucci, S. Catalini, L. Foglia, R. Cucini, F. Döring, A. Kubec, F. Koch, F. Bencivenga, A. Al Haddad, A. Gessini, A. A. Maznev, C. Cirelli, S. Gerber, B. Pedrini, G. F. Mancini, E. Razzoli, M. Burian, H. Ueda, G. Pamfilidis, E. Ferrari, Y. Deng, A. Mozzanica, P. J. M. Johnson, D. Ozerov, M. G. Izzo, C. Bottari, C. Arrell, E. J. Divall, Serhane Zerdane, M. Sander, G. Knopp, P. Beaud, H. T. Lemke, C. J. Milne, C. David, R. Torre, M. Chergui, K. A. Nelson, C. Masciovecchio, U. Staub, L. Patthey, C. Svetina, Hard X-ray transient grating spectroscopy on bismuth germanate, Nat. Photonics (2021). https://doi.org/10.1038/s41566-021-00797-9 C. Bacellar, D. Kinschel, O. Cannelli, B. Sorokin, T. Katayama, G. F. Mancini, J. R. Rouxel, Y. Obara, J. Nishitani, H. Ito, T. Ito, N. Kurahashi, C. Higashimura, S. Kudo, C. Cirelli, G. Knopp, K. Nass, P. J. M. Johnson, A. Wach, J. Szlachetko, F. A. Lima, C. J. Milne, M. Yabashi, T. Suzuki, K. Misawac, M. Chergui, "Femtosecond X-ray spectroscopy of haem proteins", Faraday Discuss., Advance Article (2021). -

Characterization of Visceral and Subcutaneous Adipose Tissue
J. Perinat. Med. 2016; 44(7): 813–835 Shali Mazaki-Tovi*, Adi L. Tarca, Edi Vaisbuch, Juan Pedro Kusanovic, Nandor Gabor Than, Tinnakorn Chaiworapongsa, Zhong Dong, Sonia S. Hassan and Roberto Romero* Characterization of visceral and subcutaneous adipose tissue transcriptome in pregnant women with and without spontaneous labor at term: implication of alternative splicing in the metabolic adaptations of adipose tissue to parturition DOI 10.1515/jpm-2015-0259 groups (unpaired analyses) and adipose tissue regions Received July 27, 2015. Accepted October 26, 2015. Previously (paired analyses). Selected genes were tested by quantita- published online March 19, 2016. tive reverse transcription-polymerase chain reaction. Abstract Results: Four hundred and eighty-two genes were differ- entially expressed between visceral and subcutaneous Objective: The aim of this study was to determine gene fat of pregnant women with spontaneous labor at term expression and splicing changes associated with par- (q-value < 0.1; fold change > 1.5). Biological processes turition and regions (visceral vs. subcutaneous) of the enriched in this comparison included tissue and vascu- adipose tissue of pregnant women. lature development as well as inflammatory and meta- Study design: The transcriptome of visceral and abdomi- bolic pathways. Differential splicing was found for 42 nal subcutaneous adipose tissue from pregnant women at genes [q-value < 0.1; differences in Finding Isoforms using term with (n = 15) and without (n = 25) spontaneous labor Robust Multichip Analysis scores > 2] between adipose was profiled with the Affymetrix GeneChip Human Exon tissue regions of women not in labor. Differential exon 1.0 ST array. Overall gene expression changes and the dif- usage associated with parturition was found for three ferential exon usage rate were compared between patient genes (LIMS1, HSPA5, and GSTK1) in subcutaneous tissues. -

Electrophysiology Read-Out Tools for Brain-On-Chip Biotechnology
micromachines Review Electrophysiology Read-Out Tools for Brain-on-Chip Biotechnology Csaba Forro 1,2,†, Davide Caron 3,† , Gian Nicola Angotzi 4,†, Vincenzo Gallo 3, Luca Berdondini 4 , Francesca Santoro 1 , Gemma Palazzolo 3,* and Gabriella Panuccio 3,* 1 Tissue Electronics, Fondazione Istituto Italiano di Tecnologia, Largo Barsanti e Matteucci, 53-80125 Naples, Italy; [email protected] (C.F.); [email protected] (F.S.) 2 Department of Chemistry, Stanford University, Stanford, CA 94305, USA 3 Enhanced Regenerative Medicine, Fondazione Istituto Italiano di Tecnologia, Via Morego, 30-16163 Genova, Italy; [email protected] (D.C.); [email protected] (V.G.) 4 Microtechnology for Neuroelectronics, Fondazione Istituto Italiano di Tecnologia, Via Morego, 30-16163 Genova, Italy; [email protected] (G.N.A.); [email protected] (L.B.) * Correspondence: [email protected] (G.P.); [email protected] (G.P.); Tel.: +39-010-2896-884 (G.P.); +39-010-2896-493 (G.P.) † These authors contributed equally to this paper. Abstract: Brain-on-Chip (BoC) biotechnology is emerging as a promising tool for biomedical and pharmaceutical research applied to the neurosciences. At the convergence between lab-on-chip and cell biology, BoC couples in vitro three-dimensional brain-like systems to an engineered microfluidics platform designed to provide an in vivo-like extrinsic microenvironment with the aim of replicating tissue- or organ-level physiological functions. BoC therefore offers the advantage of an in vitro repro- duction of brain structures that is more faithful to the native correlate than what is obtained with conventional cell culture techniques. -
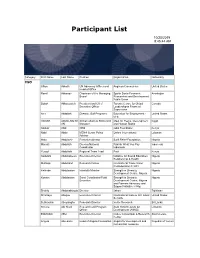
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Fall 2017 Wayne State University College of Engineering
BIOMEDICAL ENGINEERING Abu-Shaban, Kamil U. Hasan, Sazid M. Paige, Donovan J. Afework, Awet J. Heilig, Taylor C. Palani, Keerthana Alhamdani, Ahmed M. Huq, Munajj Potukutchi, Shreya Apil, Ashley C. Ismail, Omar A. Prasad, Monica Bakkar, Aya Z. John, Evangeline A. Pugh, Bradford J. Banaszak, Sean John, Rebecca Reichenbach, Grant J. Bojnowski, Jeremy J. Khan, Nabeel Saleh, Nour M. Brichacek, Brooke M. Kmiec, Michael D. Saliganan, Dan Christopher M. Bui, Krista Kulkarni, Isha S. Simard, Alexandre P. Burgin, Emma R. Labash, Kaitlyn Sodhi, Darpan K. Burton, Timothy K. Laurain, Cooper B. Stabler, Paige C. Chalhoub, Robert N. Majalikar, Yashashree Stajniak, Molly E. Dabboussi, Bilal S. Melcher, Elizabeth A. Swiantek, Travis Demeulemeester, Jake A. Mirza, Umme Aiman Syed, Ayesha Z. Diop, Fatimata S. Mora, Anthony R. Taleb, Mallak H. Donnelly, Patricia K. Mostafa, Rasik Talreja, Raghav Eck, Joshua D. Mourad, Jade Tes, David Foley, Sierra A. Mulka, Adam F. VanPaepeghem, Jonathan P. Fontana, Alessia M. Nicholson, Corey J. Weller, Noah W. Forta, Shmuel Y. Nicolas, Michael J. Wong, Sienna Hadi, Israa H. Nowak, Nicole Yzeiri, Arbora Harmer, Lance R. Omerovic, Almasa DEAN’S LIST – FALL 2017 WAYNE STATE UNIVERSITY COLLEGE OF ENGINEERING CHEMICAL ENGINEERING AND MATERIALS SCIENCE Almohri, Sayed Ahmad J. Gryko, Aleksandra Siddiqui, Abdurrafay Al-Saloum, Saja Gupta, Saloni Sidhu, Jasmine K. Banoon, Hawraa A. Isho, Simon M. Stockman, Christopher E. Butler, Colin G. Jairazbhoy, Nazir A. Taha, Daud Cormier, Emily A. Malecki, Stephanie E. Tyrrell, Kathryn J. Darrow, Margaret A. Mangulabnan, Ryan Varga, Elyse C. Ellis, Eric B. Maxey, Emily R. Veerappan, Asvin Gabrion, Charles M. Mims, Tristan X. -

PETITION List 04-30-13 Columns
PETITION TO FREE LYNNE STEWART: SAVE HER LIFE – RELEASE HER NOW! • 1 Signatories as of 04/30/13 Arian A., Brooklyn, New York Ilana Abramovitch, Brooklyn, New York Clare A., Redondo Beach, California Alexis Abrams, Los Angeles, California K. A., Mexico Danielle Abrams, Ann Arbor, Michigan Kassim S. A., Malaysia Danielle Abrams, Brooklyn, New York N. A., Philadelphia, Pennsylvania Geoffrey Abrams, New York, New York Tristan A., Fort Mill, South Carolina Nicholas Abramson, Shady, New York Cory a'Ghobhainn, Los Angeles, California Elizabeth Abrantes, Cambridge, Canada Tajwar Aamir, Lawrenceville, New Jersey Alberto P. Abreus, Cliffside Park, New Jersey Rashid Abass, Malabar, Port Elizabeth, South Salma Abu Ayyash, Boston, Massachusetts Africa Cheryle Abul-Husn, Crown Point, Indiana Jamshed Abbas, Vancouver, Canada Fadia Abulhajj, Minneapolis, Minnesota Mansoor Abbas, Southington, Connecticut Janne Abullarade, Seattle, Washington Andrew Abbey, Pleasanton, California Maher Abunamous, North Bergen, New Jersey Andrea Abbott, Oceanport, New Jersey Meredith Aby, Minneapolis, Minnesota Laura Abbott, Woodstock, New York Alexander Ace, New York, New York Asad Abdallah, Houston, Texas Leela Acharya, Toronto, Canada Samiha Abdeldjebar, Corsham, United Kingdom Dennis Acker, Los Angeles, California Mohammad Abdelhadi, North Bergen, New Jersey Judith Ackerman, New York, New York Abdifatah Abdi, Minneapolis, Minnesota Marilyn Ackerman, Brooklyn, New York Hamdiya Fatimah Abdul-Aleem, Charlotte, North Eddie Acosta, Silver Spring, Maryland Carolina Maria Acosta, -

Meeting of the Board of Trustees of the William Paterson University of New Jersey
MEETING OF THE BOARD OF TRUSTEES OF THE WILLIAM PATERSON UNIVERSITY OF NEW JERSEY Friday, March 23, 2007 The meeting was called to order at approximately 9:10 a.m. in the College Hall Board Room. BOARD MEMBERS PRESENT: Mr. Adzima, Mr. Campbell, Dr. Fan, Mr. Gruel, Mr. Mazzola, Mr. Pesce, Dr. Pruitt, Mr. Taylor, and President Speert ABSENT: Ms. Ellis, Mr. Jackson, Ms. Olmos, Ms. Temple OTHERS PRESENT: Provost and Senior Vice President Weil, Vice President Bolyai, Vice President Deller, Vice President Martone, DAG Cheryl Clarke, Dr. Schaeffer, Mrs. Santaniello, administrators, faculty, and others. ANNOUNCEMENT CONCERNING ADEQUATE NOTICE OF MEETING: In accordance with the “Open Public Meetings Act,” the Chairperson publicly announced and had entered into the minutes that “adequate notice” of this meeting was provided. In compliance with the Statute, this notice was posted on the University’s web page and distributed to The Herald News, The Record, and The Star Ledger more than 48 hours prior to this meeting. It was moved and seconded to adopt the following resolution: 3-07-1 - RESOLUTION, TO MOVE INTO EXECUTIVE SESSION (Appendix 1) The resolution was unanimously adopted. At approximately 10:55 a.m., the Public Session resumed. MINUTES OF THE DECEMBER 2, 2006, DECEMBER 8, 2006, AND JANUARY 19, 2007 BOARD MEETINGS: The minutes of the December 2, 2006, December 8, 2006 and January 19, 2007 minutes were approved with the corrections noted to the December 8, 2006 minutes, which included correcting the ranks of Drs. Ian Marshall and Michael Innis-Jimenez from Instructors to Assistant Professors and inserting the correct managerial reappointment list attachment in the minutes.