High Performance Reactive Blends Composed of Poly(P-Phenylene Sulfide)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development of Polysulfone Hollow Fiber Porous Supports for High Flux Composite Membranes: Air Plasma and Piranha Etching
fibers Article Development of Polysulfone Hollow Fiber Porous Supports for High Flux Composite Membranes: Air Plasma and Piranha Etching Ilya Borisov 1,*, Anna Ovcharova 1,*, Danila Bakhtin 1, Stepan Bazhenov 1, Alexey Volkov 1, Rustem Ibragimov 2, Rustem Gallyamov 2, Galina Bondarenko 1, Rais Mozhchil 3, Alexandr Bildyukevich 4 and Vladimir Volkov 1 1 A.V. Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, Moscow 119991, Russia; [email protected] (D.B.); [email protected] (S.B.); [email protected] (A.V.); [email protected] (G.B.); [email protected] (V.V.) 2 Kazan National Research Technological University, Kazan 420015, Russia; [email protected] (R.I.); [email protected] (R.G.) 3 National Research Nuclear University “MEPhI”, Moscow 115409, Russia; [email protected] 4 Institute of Physical Organic Chemistry, National Academy of Sciences of Belarus, Minsk 220072, Belarus; [email protected] * Correspondence: [email protected] (I.B.); [email protected] (A.O.); Tel.: +7-495-955-4893 (I.B.); +7-495-647-59-27 (A.O.) Academic Editors: Alberto Figoli and Tao He Received: 30 December 2016; Accepted: 4 February 2017; Published: 13 February 2017 Abstract: For the development of high efficiency porous supports for composite membrane preparation, polysulfone (PSf) hollow fiber membranes (outer diameter 1.57 mm, inner diameter 1.12 mm) were modified by air plasma using the low temperature plasma treatment pilot plant which is easily scalable to industrial level and the Piranha etch (H2O2 + H2SO4). Chemical and plasma modification affected only surface layers and did not cause PSf chemical structure change. -

Radel® PPSU, Udel® PSU, Veradel® PESU & Acudel® Modified PPSU
Radel ® | Udel ® | Veradel ® | Acudel ® Radel® PPSU, Udel® PSU, Veradel® PESU & Acudel® modified PPSU Processing Guide SPECIALT Y POLYMERS 2 \ Sulfone Polymers Processing Guide Table of Contents Introduction ............................. 5 Part Ejection . 14 Draft . 14 Ejector pins and/or stripper plates . 14 Sulfone Polymers........................ 5 Udel® Polysulfone (PPSU) . 5 Injection Molding Equipment ............. 15 ® Veradel Polyethersulfone (PESU) . 5 Controls . 15 ® Radel Polyphenylsulfone (PPSU) . 5 Clamp . 15 ® Acudel modified PPSU . 5 Barrel Capacity . 15 Press Maintenance . 15 Resin Drying . .6 Screw Design . 15 Rheology................................ 8 Screw Tips and Check Valves . 15 Viscosity-Shear Rate ..................... 8 Nozzles . 16 Molding Process . 16 Resin Flow Characteristics . 9 Melt flow index . 9 Polymer Injection or Mold Filling . 16 Spiral flow . 9 Packing and Holding . 17 Injection Molding . .10 Cooling . 17 Molds and Mold Design .................. 10 Machine Settings ....................... 17 Tool Steels . 10 Barrel Temperatures . 17 Mold Dimensions . 10 Mold Temperature . 18 Mold Polishing . 10 Residence Time in the Barrel . 18 Mold Plating and Surface Treatments . 10 Injection Rate . 18 Tool Wear . 10 Back Pressure . 18 Mold Temperature Control . 10 Screw Speed . 18 Mold Types . 11 Shrinkage . 18 Two-plate molds . 11 Three-plate molds . 11 Regrind ............................... 19 Hot runner molds . 11 Cavity Layout . 12 Measuring Residual Stress ............... 19 Runner Systems . 12 Extrusion............................... 22 Gating . 12 Sprue gating . 12 Edge gates . 13 Predrying ............................. 22 Diaphragm gates . 13 Tunnel or submarine gates . 13 Extrusion Temperatures ................. 22 Pin gates . 13 Screw Design Recommendations . 22 Gate location . 13 Venting . 14 Sulfone Polymers Processing Guide / 3 Die Design ............................. 22 Extruded Product Types . 23 Wire . 23 Film . 23 Sheet . 23 Piping and tubing . 23 Start-Up, Shut-Down, and Purging ....... -

Technical Datasheet: Udel® P-1700
Udel® P-1700 Polysulfone Solvay Specialty Polymers www.ulprospector.com Technical Data Product Description Udel® P-1700 polysulfone (PSU) is a tough, rigid, high-strength thermoplastics suitable for continuous use up to 300°F (149°C). It is resistant to oxidation and hydrolysis and withstand prolonged exposure to high temperatures and repeated sterilization. Udel® P-1700 polysulfone is highly resistant to mineral acids, alkali and salt solutions. Resistance to detergents and hydrocarbon oils is good, but the resin may be attacked by polar solvents such as ketones, chlorinated hydrocarbons and aromatic hydrocarbons. These resins are also highly resistant to degradation by gamma or electron beam radiation. Electrical properties of Udel® P-1700 polysulfones are stable over a wide temperature range and after immersion in water or exposure to high humidity. The resins comply with FDA 21 CFR 177.1655 and may be used in articles intended for repeated use in contact with foods. Additionally, they are approved by the NSF, by the Department of Agriculture for contact with meat and poultry and by the 3-A Sanitary Standards of the Dairy Association. • Transparent: Udel® P-1700 CL 2611 CMP • Transparent: Udel® P-1700 NT 06 • Transparent: Udel® P-1700 NT 11 • Opaque Black : Udel® P-1700 BK 937 • Opaque White: Udel® P-1700 WH 6417 • Opaque White: Udel® P-1700 WH 7407 • Opaque Gray: Udel® P-1700 GY 8057 General Material Status • Commercial: Active Literature 1 • Technical Datasheet UL Yellow Card 2 • E36098-231084 • Solvay Specialty Polymers Search for -

Studies on Radiation Crosslinking of Polysulfone
JP0050427 JAERI-Conf 2000-001 Studies on Radiation Crosslinking of Polysulfone Xiaoguang Zhong Jiazhen Sun Changchun Institute of Applied Chemistry Chinese Academy of Sciences Changchun 130022, China Polysulfone is a kind of high temperature-resistance and radiation-resistance engineering plastic. The chemical structure is as follows: Brown (1), Lyon (2), Sasuga (3), et al have already studied its radiation effect. We studied CH3 radiation crosslinking effect of polysulfone by using of XPS, ESR, and CG methods and got some new results. Results and Discussions 1. Study radiation crosslinking of polysulfone by XPS method Because of conjugate system of benzene ring, polymer material which contains of benzene ring will appear shake-up peak in XPS spectra. Wanxi Zhang (4) shows that during radiation crosslinking of polystyrene increases with radiation dose and the intensity of shake-up peak decreases gradually with increase of radiation dose and crosslinking degree. This suggests that radiation crosslinking destroyed conjugate system of benzene ring. During radiation crosslinking of polysulfone, we find rules of shake-up peaks in the XPS spectra are different at different radiation crosslinking temperature. At lower temperature the intensity of shake-up peak decreases with the increase of radiation dose. This rule is similar to that of radiation crosslinking of polystyrene. The results are shown in Fig. 1. 183- JAERI-Conf 2000-001 Fig. 1. The spectra of radiation crosslinking of polysulfone at 70"C Comparing with radiation crosslinking at lower temperature, the intensity of shake-up peak increases with radiation dose when radiation crosslinking reaction takes place at temperature above glass transition temperature of polysulfone. -

US3636140.Pdf
Jan. 18, 1972 A. F. INGULLI ETA 3,636,140 THERMOPLASTIC RESIN BLEND OF POLY SULFONE WITH ABS Filed Aug. 4, 1969 5. Sheets-Sheet, 2. R O /O 20 30 40 0 60 22 80 20 M22 (7, AOA. Yuo/MAOM/A /W AA S adze wa /MVA MV7 OAS Air 7- 4. a Zafa AA A. MM/G/ZZ / A/a. Mary A. at 7A air 2- 9% agew, Jan. 18, 1972 A. F. NGULL ET All- 3,636,140 "I HERMOPLASTIC RESIN BLEND OF POLYSULFONE WITH ABS Filed Aug. 4, 1969 5. Sheets-Sheet 3 S | | | | | | \ / | | | | |\ 1 \ | | | | | | | | \ | | | | | | | N TTTTTTTT O 3O 20 O SO 70 so 90 /OO 7. AozYuva /owa w Aaj azawo 2g z 7-5 MM/a M72A S. AAAAAA M. MMMGO/4 AM AyAAMA 1 A... a A. JAA larus 9.4 AG AW 7 Jan. 18, 1972 A. F. NGULL FT All- 3,636,140 TERMOPLASTIC RESIN BLEND OF POLYSULFONE WITH ABS Filed Aug. 4, 1969 5 Sheets-Sheet 4. 24.0 -- -T 22.ol | | | || 20.0 I Y Mas t SR I W. n w /4.0 | -- N - S v 2.0 HI N | n N S s S. v O /O 20 30 10 60 60 70 &0 70 /00 7./oz ruz/z/-owa //v4 as aza/v4 MM VAM/OA 27zz7 a AAAMAA A. MM6t/44/ A/AMMA)1 A. az Zafa 9ers /.4- a 47AM/7 Jan. 18, 1972 A. F. NGULL ET All- 3,636,140 THERMOPLASTIC RESIN BLEND OF POLYSULFONE WITH ABS Filed Aug. 4, 1969 5 Sheets-Sheet 5 JO J00 290 28 O 270 28 O 25 O 24 O 2.3 O O O 20 JO 40 50 60 70 8 O 3 O LOO % AOZyura/4 MoM2 //v 44, aza/V2 , , , . -
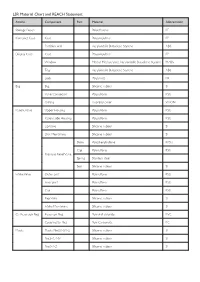
LSR Material Chart and REACH Statement
LSR Material Chart and REACH Statement Article Component Part Material Abbreviation Storage Pouch Polyethylene PE Compact Case Case Polypropylene PP Partition wall Acrylonitrile Butadiene Styrene ABS Display Case Case Polypropylene PP Window Methyl Methacrylate Acrylonitrile Butadiene Styrene MABS Tray Acrylonitrile Butadiene Styrene ABS Lock Polyamide PA Bag Bag Silicone rubber SI Valve Connector Polysulfone PSU O-Ring Fluorelastomer VITON Patient Valve Upper Housing Polysulfone PSU Patient side Housing Polysulfone PSU Lip Valve Silicone rubber SI Disk Membrane Silicone rubber SI Stem Polyphenylsulfone PPSU Cap Polysulfone PSU Pressure Relief Valve Spring Stainless steel Seal Silicone rubber SI Intake Valve Outer part Polysulfone PSU Inner part Polysulfone PSU Cap Polysulfone PSU Flap Valve Silicone rubber SI Intake Membrane Silicone rubber SI O² Reservoir Bag Reservoir Bag Polyvinyl chloride PVC Coupling for Bag Poly Carbonate PC Masks Masks No.00-0/1-2 Silicone rubber SI No.3-4, 4-5+ Silicone rubber SI No.0-1-2 Silicone rubber SI Article Component Part Material Abbreviation Mask Cover Polysulfone PSU Lock Clip Stainless steel Head Strap w/Ring Strap Thermoplastic Elastomer TPE Attachment Ring Polysulfone PSU Expiration Diverter Housing Polysulfone PSU Center gasket Silicone rubber SI External gasket Silicone rubber SI Extension Tube Tube Silicone rubber SI Coupling Polysulfone PSU Manometer Polysulfone PSU Connector Hanging Loop Silicone rubber SI Wall Mount Acrylonitrile Butadiene Styrene ABS Wall Bracket PolyOxyMethylene POM Laerdal Statement for REACH Regulation EC No 1907/2006 Substances of Very High Concern are not used in concentrations above 0.1% (w/w) in the production of all models of the Laerdal Silicone Resuscitator. -

Polysulfone PSU1000 Datasheet
Quality Plastics Since 1936 POLYSULFONE - PSU1000 Characteristics * Broad Temperature Range Capability - Hot Water and Steam Performance to 300º F (150 C) * Good Thermal and Electrical Insulation Characteristics * Hydrolysis Resistant * Radiation Stability * Low Ionic Impurity Description PSU 1000 Polysulfone is an amber semi-transparent, heat resistant, high performance engineering thermoplastic. It offers excellent mechanical, electrical and improved chemical resistance properties relative to polycarbonate. Polysulfone's properties remain relatively consistent over a broad temperature range, from -150º to 300º F (-100º to 150º C). PSU Polysulfone offers high chemical resistance to acidic and salt solutions, and good resistance to detergents, hot water and steam. In addition, polysulfone has excellent radiation stability and offers low ionic impurity levels. PSU 1000 Polysulfone often replaces polycarbonate when higher temperatures, improved chemical resistance or autoclavability is required. It is commonly used for analytical instrumentation, medical devices and semiconductor process equipment components. Forms Available ROD .250 to 6.00 " diameter, Lengths to 12 feet TUBE Custom produced as requested PLATE .250" to 3.000" thick, Widths to 24", Lengths to 48" COLOR Amber Typical Property Values Polysulfone MECHANICAL @ 73ºF PSU-1000 Specific Gravity 1.24 Tensile Strength psi 10,200 Tensile Modulus of Elasticity psi 390,000 Tensile Elongation ( at Break ) % 30 Flexural Strength psi 15,000 Flexural Modulus of Elasticity psi 400,000 Shear Strength psi 9,000 Compressive Strength, 10% Deformation psi 13,000 Compressive Modulus of Elasticity psi 375,000 Rockwell Hardness M Scale 82 Izod Impact Strength, Notched ft-lbs/in. of notch 1.30 Coefficient of Friction, Dynamic ( Dry vs. Steel ) Limiting PV ( 4 :1 Safety Factor Applied ) ft.lbs/in.² min Wear Factor in³-min/ft.lbs. -

Dissertation Diógenes Rojas
Poly (styrene - co - maleic anhydride) and Polystyrene Grafted with Poly(ether amines): Synthesis, Characterization and Gas Separation Performance Dissertation zur Erlangung des akademischen Grades Doktor der Naturwissenschaften (Dr. rer. nat) der Technischen Fakultät der Christian-Albrechts-Universität zu Kiel Diógenes Rojas Kiel 2010 Poly (styrene - co - maleic anhydride) and Polystyrene Grafted with Poly(ether amines): Synthesis, Characterization and Gas Separation Performance Dissertation zur Erlangung des akademischen Grades Doktor der Naturwissenschaften (Dr. rer. nat) der Technischen Fakultät der Christian-Albrechts-Universität zu Kiel Diógenes Rojas Kiel 2010 1. Gutachter Prof. Dr. Volker Abetz 2. Gutachter Prof. Dr. Klaus Rätzke Datum der mündlichen Prüfung 15.11.2010 Acknowledgement After a difficult but satisfactory challenge to complete my Ph.D. I would like to express my gratitude towards my supervisor Prof. Dr. Peter Simon. His kindness, suggestions, and observations have been an enormous contribution to my project; I hope to have a chance to continue in this productive exchange of ideas and experiences in the future. I am also thankful to Dr. Luis Antonio de Almeida Prado for the collaboration and fruitful discussion of ideas generating a better and qualified work. Special thanks go to my co-workers at GKSS- Institute of Polymer Research (Helmholtz-Zentrum Geesthacht) for having been helpful and contributing to my work; namely, Susanne Novak, Maren Brinkmann, Karen-Marita Prause, Silvio Neumann, Carsten Scholles and Brigitte Lademann. I also thank Dr. Wilfredo Yave and Dr. Carmen Nistor for the abundant information and discussion about the gas separation membrane. Furthermore, I am grateful to Connie Kampmann to have been a great spiritual support during my journey to complete my Ph.D. -

Ether Ether Ketone): Monitoring the Meta-Fluorine Displacement in 3,5,4’- Trifluorobenzophenone
Wright State University CORE Scholar Browse all Theses and Dissertations Theses and Dissertations 2017 Toward The Synthesis of Functionalized Poly (Ether Ether Ketone): Monitoring the meta-Fluorine Displacement in 3,5,4’- trifluorobenzophenone Giovanni Covarrubias Wright State University Follow this and additional works at: https://corescholar.libraries.wright.edu/etd_all Part of the Chemistry Commons Repository Citation Covarrubias, Giovanni, "Toward The Synthesis of Functionalized Poly (Ether Ether Ketone): Monitoring the meta-Fluorine Displacement in 3,5,4’-trifluorobenzophenone" (2017). Browse all Theses and Dissertations. 1725. https://corescholar.libraries.wright.edu/etd_all/1725 This Thesis is brought to you for free and open access by the Theses and Dissertations at CORE Scholar. It has been accepted for inclusion in Browse all Theses and Dissertations by an authorized administrator of CORE Scholar. For more information, please contact [email protected]. TOWARD THE SYNTHESIS OF FUNCTIONALIZED, SEMI-CRYSTALLINE POLY (ETHER ETHER KETONE): MONITORING THE META-FLUORINE DISPLACEMENT IN 3,5,4’-TRIFLUOROBENZOPHENONE A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By: GIOVANNI COVARRUBIAS B.S. Loras College, 2013 2017 Wright State University WRIGHT STATE UNIVERSITY GRADUATE SCHOOL May 18th, 2017 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY Giovanni Covarrubias ENTITLED Toward The Synthesis of Functionalized Poly (Ether Ether Ketone)s: Monitoring the meta-Fluorine Displacement in 3,5,4’-trifluorobenzophenone BE ACCEPTED IN PARTIAL FULLFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science. ____________________________ Eric Fossum, Ph. D. Thesis Advisor ____________________________ David Grossie, Ph. D. Chair, Department of Chemistry Committee on Final Examination _____________________________ Eric Fossum, Ph. -

(12) United States Patent (10) Patent No.: US 8,211,455 B2 Schwarz (45) Date of Patent: *Jul
USOO821 1455B2 (12) United States Patent (10) Patent No.: US 8,211,455 B2 Schwarz (45) Date of Patent: *Jul. 3, 2012 (54) IMPLANTABLE OR INSERTABLE MEDICAL 6,527,938 B2 * 3/2003 Bales et al. ................... 205/229 DEVICES FOR CONTROLLED DELIVERY OF 6,558,686 B1* 5/2003 Darouiche ... ... 424/423 6,558,809 B1* 5/2003 Kelch et al. ... 428,520 ATHERAPEUTICAGENT 6,630,215 B1 * 10/2003 Oda et al. ..................... 428, 35.7 2002fOO16574 A1 2/2002 Wang et al. (75) Inventor: Marlene C. Schwarz, Auburndale, MA 2002/0107330 A1 8/2002 Pinchuk et al. ............... 525,242 (US) 2004/01754.06 A1* 9, 2004 Schwarz ....................... 424/423 (73) Assignee: Boston Scientific Scimed, Inc., Maple FOREIGN PATENT DOCUMENTS Grove, MN (US) EP O 166998 A2 * 1, 1986 EP O166998 1, 1986 (*) Notice: Subject to any disclaimer, the term of this wo WO ...i. A2 SES patent is extended or adjusted under 35 WO WO98,563.12 A1 * 12, 1998 U.S.C. 154(b) by 774 days. WO WOOO,21584 4/2000 WO WOOOf 62830 10, 2000 This patent is Subject to a terminal dis- WO WO O2/43788 A2 6, 2002 claimer. OTHER PUBLICATIONS (21) Appl. No.: 10/175,136 Odian Principles of Polymerization 3rd ed., 1991, John Wiley and Sons, p. 142.* (22) Filed: Jun. 19, 2002 O. Perez-Camacho et al., “Preparation of Hydroxyl-Functionalized SEBS for in Situ Graft Reaction Compatibilizing Agents.” Journal of (65) Prior Publication Data Applied Polymer Science, 64(13): 2519-2528 (1997). US 2003/0236514 A1 Dec. 25, 2003 Gantrez. -

Process for Preparing Isotropic Microporous Polysulfone Membranes
Europa,schesP_ MM M M M MM Ml II II II II II I II J European Patent Office i-oo n * © Publication number: 0 362 588 B1 Office europeen* des.. brevets , © EUROPEAN PATENT SPECIFICATION © Date of publication of patent specification: 09.11.94 © Int. CI.5: B01 D 69/08, B01D 69/06, B01D 67/00, B01D 71/68 © Application number: 89117000.3 @ Date of filing: 14.09.89 © Process for preparing isotropic microporous polysulfone membranes. ® Priority: 23.09.88 US 248880 hollow fibers. II. Morphology" @ Date of publication of application: © Proprietor: W.R. GRACE & CO. 11.04.90 Bulletin 90/15 Grace Plaza 1114 Avenue of the Americas © Publication of the grant of the patent: New York New York 10036 (US) 09.11.94 Bulletin 94/45 @ Inventor: Ly, Ann L. © Designated Contracting States: 14 Sheridan Road DE FR GB IT Bedford MA 01730 (US) Inventor: Chu, Chaokang © References cited: 6 Wildwood Road EP-A- 0 086 365 Lexington MA 02173 (US) EP-A- 0 168 783 Inventor: Nguyen, Thanh D. DE-A- 3 342 824 28 Manning Street US-A- 4 051 300 Billerica MA 01821 (US) US-A- 4 612 119 INDUSTRIAL & ENGINEERING CHEMISTRY © Representative: UEXKULL & STOLBERG Paten- RESEARCH, vol. 26, no. 11, November 1987, tanwalte pages 2385-2389, American Chemical Soci- Beselerstrasse 4 ety,Washington, DC, US; L.Y. LAFRENIERE et D-22607 Hamburg (DE) al.:"Effect of polyvinylpyrrolidone additive on 00 the performance of polyethersulfone ultrafil- 00 tration membranes" 00 m Journal of applied polymer science, Vol.21, CM 1977, pp.1 65-80; Cabasso et al., "Polysulfone CO 00 Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Polymer Properties and Classification
Polymer Characterization LDPE Polyethylene low density HDPE Polyethylene high density ABS Acrylonitrile-butadiene-styrene SAN Styrene-acrylonitrile copolymer EVA Polyethylene co-vinyl acetate PVA Polyvinyl acetate PerkinElmer Solutions for Polymer Characterization Tg(ºC): -130 to 100 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): -130 to 100 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): 110 to 125 CpJ/(g*K): 1,25 to 1,7 Tg(ºC): 95 to 110 CpJ/(g*K): 1,2 Tg(ºC): -45 to 20 CpJ/(g*K): 2,3 Tg(ºC): 25 to 35 CpJ/(g*K): - Tm(ºC): 100 to 120 DHf (J/g): - Tm(ºC): 130 to 140 DHf (J/g): 293 Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): 30 to 100 DHf (J/g): 10 to 100 Tm( ºC ): - DHf (J/g): - Td(ºC): 490 to 500 Td(ºC): 490 to 500 Td(ºC): 420 Td(ºC): 420 Td(ºC): 480 Td(ºC): - PP Polypropylene PS PMMA Polymethylmethacrylate PBMA CA Polystyrene Polybuthylmethacrylate Cellulose acetate EP Epoxy resin Molecular Spectroscopy FTIR Differential Scanning Calorimetry Tg(ºC): -20 to -5 CpJ/(g*K): 1,8 Tg(ºC): 90 to 110 Cp (J/g*K): 1,8 to 3,4 Tg(ºC): 85 to 100 CpJ/(g*K): 1,45 to 1,5 Tg(ºC): 15 to 25 CpJ/(g*K): - Tg(ºC): 45 to 60 CpJ/(g*K): - Tg(ºC): 50 to 200 CpJ/(g*K): 1,6 to 2,1 Identify and quantitate organic molecules and compounds, Glass transition & melting temperatures, crystallinity, heat of Understand chemical & physical composition of laminates & fusion, reaction rates, specific heat & heat capacity, curing, Tm(ºC): 165 to 175 DHf (J/g): 207 Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm(ºC): - DHf (J/g): - Tm( ºC): - DHf (J/g): - adhesives , Troubleshoot