Sectm1a Deficiency Aggravates Inflammation-Triggered Cardiac Dysfunction Through Disruption of Lxrα Signaling in Macrophages
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic and Expression Profiling of Chromosome 17 in Breast Cancer Reveals Complex Patterns of Alterations and Novel Candidate Genes
[CANCER RESEARCH 64, 6453–6460, September 15, 2004] Genomic and Expression Profiling of Chromosome 17 in Breast Cancer Reveals Complex Patterns of Alterations and Novel Candidate Genes Be´atrice Orsetti,1 Me´lanie Nugoli,1 Nathalie Cervera,1 Laurence Lasorsa,1 Paul Chuchana,1 Lisa Ursule,1 Catherine Nguyen,2 Richard Redon,3 Stanislas du Manoir,3 Carmen Rodriguez,1 and Charles Theillet1 1Ge´notypes et Phe´notypes Tumoraux, EMI229 INSERM/Universite´ Montpellier I, Montpellier, France; 2ERM 206 INSERM/Universite´ Aix-Marseille 2, Parc Scientifique de Luminy, Marseille cedex, France; and 3IGBMC, U596 INSERM/Universite´Louis Pasteur, Parc d’Innovation, Illkirch cedex, France ABSTRACT 17q12-q21 corresponding to the amplification of ERBB2 and collinear genes, and a large region at 17q23 (5, 6). A number of new candidate Chromosome 17 is severely rearranged in breast cancer. Whereas the oncogenes have been identified, among which GRB7 and TOP2A at short arm undergoes frequent losses, the long arm harbors complex 17q21 or RP6SKB1, TBX2, PPM1D, and MUL at 17q23 have drawn combinations of gains and losses. In this work we present a comprehensive study of quantitative anomalies at chromosome 17 by genomic array- most attention (6–10). Furthermore, DNA microarray studies have comparative genomic hybridization and of associated RNA expression revealed additional candidates, with some located outside current changes by cDNA arrays. We built a genomic array covering the entire regions of gains, thus suggesting the existence of additional amplicons chromosome at an average density of 1 clone per 0.5 Mb, and patterns of on 17q (8, 9). gains and losses were characterized in 30 breast cancer cell lines and 22 Our previous loss of heterozygosity mapping data pointed to the primary tumors. -

Identification of Novel Alternative Splice Isoforms of Circulating Proteins in a Mouse Model of Human Pancreatic Cancer
Research Article Identification of Novel Alternative Splice Isoforms of Circulating Proteins in a Mouse Model of Human Pancreatic Cancer Rajasree Menon,1 Qing Zhang,3 Yan Zhang,1 Damian Fermin,1 Nabeel Bardeesy,4 Ronald A. DePinho,5 Chunxia Lu,2 Samir M. Hanash,3 Gilbert S. Omenn,1 and David J. States1 1Center for Computational Medicine and Biology and 2Pediatric Endocrinology, University of Michigan, Ann Arbor, Michigan; 3Fred Hutchinson Cancer Research Center, Seattle, Washington; and 4Center for Cancer Research, Massachusetts General Hospital; 5Center for Applied Cancer Science, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts Abstract database are scored as high, medium, or low confidence, reflecting the amount of cumulative evidence in support of the existence of a To assess the potential of tumor-associated, alternatively particular alternatively spliced sequence. Evidence is collected from spliced gene products as a source of biomarkers in biological clustering of ESTs, mRNA sequences, and gene model predictions. fluids, we have analyzed a large data set of mass spectra We modified the ECgene database to include three-frame trans- derived from the plasma proteome of a mouse model of lations of the cDNA sequences (5) to determine the occurrence of human pancreatic ductal adenocarcinoma. MS/MS spectra novel splice variant proteins. An important development in recent were interrogated for novel splice isoforms using a non- years is the substantial improvement in tandem mass spectrometry redundant database containing an exhaustive three-frame instrumentation for proteomics, allowing in-depth analysis and translation of Ensembl transcripts and gene models from confident identifications even for proteins coded by mRNA ECgene. -

Development and Validation of an Individualized Immune Prognostic Signature in Early-Stage Nonsquamous Non–Small Cell Lung Cancer
Supplementary Online Content Li B, Cui Y, Diehn M, Li R. Development and validation of an individualized immune prognostic signature in early-stage nonsquamous non–small cell lung cancer. JAMA Oncol. Published online July 6, 2017. doi:10.1001/jamaoncol.2017.1609 eMethods. Data Preprocessing, Assessment of Robustness, and Comparison With Commercialized Lung Biomarker eResults. Analysis and Robustness of IRGPs eTable 1. Details About the Data Sets Used in This Study eTable 2. Clinical and Pathologic Features of Patients in Meta-training, Meta-testing, and Independent Validation Cohorts eTable 3. Model Information About IRGPI eTable 4. Univariate and Multivariate Analyses of Prognostic Factors in Meta-training, Meta-testing, and Independent Validation Data Sets eTable 5. Biological Processes Overrepresented by Genes Consisting of IRGPI eTable 6. RMS Time Ratio Between Low- and High-Risk Groups Based on IRGPI or ICPI in Different Data Sets eFigure 1. Overview of the Construction and Validation of Immune and Composite Immune/Clinical Signatures eFigure 2. Time-Dependent ROC Curve for IRGPI in the Meta-training Data Set at 5 Years eFigure 3. Kaplan-Meier Curve of Overall Survival for Patients With Different IRGPI Risks eFigure 4. Kaplan-Meier Curve of Overall Survival for Early- and Late-Stage Patients With Different IRGPI Risks eFigure 5. Kaplan-Meier Curve of Overall Survival for Stage IA and IB Patients With Different IRGPI Risks eFigure 6. C-index Comparison Between IRGPI and 2 Existing Biomarkers eFigure 7. Time-Dependent ROC for ICPI and RMS Curve for ICPI and IRGPI in Meta-training Data Set eFigure 8. Kaplan-Meier Curves for Overall Survival of All Patients Stratified by the IRGPI and the ICPI eFigure 9. -

Cysteine‑Rich 61‑Associated Gene Expression Profile Alterations in Human Glioma Cells
MOLECULAR MEDICINE REPORTS 16: 5561-5567, 2017 Cysteine‑rich 61‑associated gene expression profile alterations in human glioma cells RUI WANG1, BO WEI2, JUN WEI3, YU TIAN2 and CHAO DU2 Departments of 1Radiology, 2Neurosurgery and 3Science and Education Section, China‑Japan Union Hospital of Jilin University, Changchun, Jilin 130033, P.R. China Received January 28, 2016; Accepted February 20, 2017 DOI: 10.3892/mmr.2017.7216 Abstract. The present study aimed to investigate gene be critical for maintaining the role of CYR61 during cancer expression profile alterations associated with cysteine‑rich 61 progression. (CYR61) expression in human glioma cells. The GSE29384 dataset, downloaded from the Gene Expression Omnibus, Introduction includes three LN229 human glioma cell samples expressing CYR61 induced by doxycycline (Dox group), and three Cysteine‑rich 61 (CYR61) is a secreted, cysteine‑rich, control samples not exposed to doxycycline (Nodox group). heparin‑binding protein (1) involved in a variety of cellular Differentially expressed genes (DEGs) between the Dox and functions including adhesion, migration and proliferation (2). Nodox groups were identified with cutoffs of |log2 fold change Previously, Xie et al (3) reported that CYR61 was overexpressed (FC)|>0.5 and P<0.05. Gene ontology and Kyoto Encyclopedia in 66 primary gliomas compared with healthy brain samples, of Genes and Genomes pathway enrichment analyses for and that CYR61 expression was significantly correlated with DEGs were performed. Protein‑protein interaction (PPI) tumor grade and patient survival (3). CYR61‑overexpressing network and module analyses were performed to identify glioma cells were observed to have an increased proliferation the most important genes. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

KDML: a Machine-Learning Framework for Inference of Multi-Scale Gene Functions from Genetic Perturbation Screens
bioRxiv preprint doi: https://doi.org/10.1101/761106; this version posted September 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. KDML: a machine-learning framework for inference of multi-scale gene functions from genetic perturbation screens Heba Z. Sailem*1,2, Jens Rittscher1,2, Lucas Pelkmans3 1 Institute of Biomedical Engineering, Department of Engineering Science, Old Road Campus Research Building, University of Oxford OX3 7DQ, UK 2 Big Data Institute, University of Oxford, Li Ka Shing Centre for Health Information and Discovery, Old Road Campus Research Building, Oxford OX3 7LF, UK 3 Department of Molecular Life Sciences, Winterthurerstrasse 190, 8057, University of Zurich, Switzerland * Address correspondence to: [email protected] Running title Mapping of phenotypes to gene functions 1 bioRxiv preprint doi: https://doi.org/10.1101/761106; this version posted September 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Characterising context-dependent gene functions is crucial for understanding the genetic bases of health and disease. To date, inference of gene functions from large- scale genetic perturbation screens is based on ad-hoc analysis pipelines involving unsupervised clustering and functional enrichment. We present Knowledge-Driven Machine Learning (KDML), a framework that systematically predicts multiple functions for a given gene based on the similarity of its perturbation phenotype to those with known function. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -
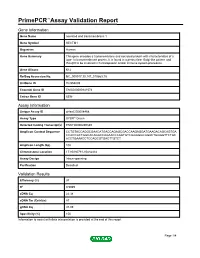
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name secreted and transmembrane 1 Gene Symbol SECTM1 Organism Human Gene Summary This gene encodes a transmembrane and secreted protein with characteristics of a type 1a transmembrane protein. It is found in a perinuclear Golgi-like pattern and thought to be involved in hematopoietic and/or immune system processes. Gene Aliases K12 RefSeq Accession No. NC_000017.10, NT_010663.15 UniGene ID Hs.558009 Ensembl Gene ID ENSG00000141574 Entrez Gene ID 6398 Assay Information Unique Assay ID qHsaCID0008468 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000269389 Amplicon Context Sequence CCTGTACCAGGCGAACATGACCAGAGCGACCAAGAGGATGAAGACAGCAGTGA CCACCGCTGGCACAGGCCAGAACCCAGTGTCGGGGGCGGACTGGGGTTCTGC ACCTGAAACCTCCAGCGTGACTTGTCT Amplicon Length (bp) 102 Chromosome Location 17:80280781-80282482 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 97 R2 0.9989 cDNA Cq 22.38 cDNA Tm (Celsius) 87 gDNA Cq 37.09 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report SECTM1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -
Tex19 and Sectm1 Concordant Molecular Phylogenies Support Co
Bianchetti et al. BMC Evolutionary Biology (2015) 15:222 DOI 10.1186/s12862-015-0506-y RESEARCH ARTICLE Open Access Tex19 and Sectm1 concordant molecular phylogenies support co-evolution of both eutherian-specific genes Laurent Bianchetti1*, Yara Tarabay2,5, Odile Lecompte3, Roland Stote1, Olivier Poch3, Annick Dejaegere1 and Stéphane Viville2,4 Abstract Background: Transposable elements (TE) have attracted much attention since they shape the genome and contribute to species evolution. Organisms have evolved mechanisms to control TE activity. Testis expressed 19 (Tex19) represses TE expression in mouse testis and placenta. In the human and mouse genomes, Tex19 and Secreted and transmembrane 1 (Sectm1) are neighbors but are not homologs. Sectm1 is involved in immunity and its molecular phylogeny is unknown. Methods: Using multiple alignments of complete protein sequences (MACS), we inferred Tex19 and Sectm1 molecular phylogenies. Protein conserved regions were identified and folds were predicted. Finally, expression patterns were studied across tissues and species using RNA-seq public data and RT-PCR. Results: We present 2 high quality alignments of 58 Tex19 and 58 Sectm1 protein sequences from 48 organisms. First, both genes are eutherian-specific, i.e., exclusively present in mammals except monotremes (platypus) and marsupials. Second, Tex19 and Sectm1 have both duplicated in Sciurognathi and Bovidae while they have remained as single copy genes in all further placental mammals. Phylogenetic concordance between both genes was significant (p-value < 0.05) and supported co-evolution and functional relationship. At the protein level, Tex19 exhibits 3 conserved regions and 4 invariant cysteines. In particular, a CXXC motif is present in the N-terminal conserved region. -

Network Mining Approach to Cancer Biomarker Discovery
NETWORK MINING APPROACH TO CANCER BIOMARKER DISCOVERY THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Praneeth Uppalapati, B.E. Graduate Program in Computer Science and Engineering The Ohio State University 2010 Thesis Committee: Dr. Kun Huang, Advisor Dr. Raghu Machiraju Copyright by Praneeth Uppalapati 2010 ABSTRACT With the rapid development of high throughput gene expression profiling technology, molecule profiling has become a powerful tool to characterize disease subtypes and discover gene signatures. Most existing gene signature discovery methods apply statistical methods to select genes whose expression values can differentiate different subject groups. However, a drawback of these approaches is that the selected genes are not functionally related and hence cannot reveal biological mechanism behind the difference in the patient groups. Gene co-expression network analysis can be used to mine functionally related sets of genes that can be marked as potential biomarkers through survival analysis. We present an efficient heuristic algorithm EigenCut that exploits the properties of gene co- expression networks to mine functionally related and dense modules of genes. We apply this method to brain tumor (Glioblastoma Multiforme) study to obtain functionally related clusters. If functional groups of genes with predictive power on patient prognosis can be identified, insights on the mechanisms related to metastasis in GBM can be obtained and better therapeutical plan can be developed. We predicted potential biomarkers by dividing the patients into two groups based on their expression profiles over the genes in the clusters and comparing their survival outcome through survival analysis. -

Identification of Important Cell Cycle Regulators And
IDENTIFICATION OF IMPORTANT CELL CYCLE REGULATORS AND NOVEL GENES IN SPECIFIC TISSUES USING MICROARRAY ANALYSIS, BIOINFORMATICS AND MOLECULAR TOOLS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Jibin Zhang Graduate Program in Animal Sciences The Ohio State University 2015 Dissertation Committee: Dr. Michael E. Davis, Advisor Dr. Kichoon Lee, Co-Advisor Dr. Macdonald Wick Dr. Harald Vaessin Copyrighted by Jibin Zhang 2015 ABSTRACT Most biological activities in animals are complicated processes regulated by expression of a large number of genes. In addition, most functions of one particular tissue are regulated not only by gene expression but also by signals delivered by other tissues, comprising an extremely complex network in an organism that is still a mystery in many aspects. Fortunately, microarray technology provides an efficient method to select the important genes involved in a complex physiological activity, as well as the novel genes belonging to a large category. Meta-analysis of various microarray studies dealing with similar problems can further provide a precise and economical way to identify critical or novel genes and predict gene functions based on comparison of gene expression. If we can further combine the meta-analysis of microarrays with various molecular technologies, it will be a powerful strategy in biological studies. Because the bioinformatic study of microarray data can reveal explicit targets and outline clear direction for molecular biological studies, whereas the molecular biological studies can in turn provide further confirmation of the target genes, reliable evidence for the predicted functions, as well as extended applications for the target genes, can be obtained from bioinformatic study of microarray data. -
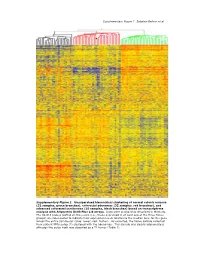
Supplementary Data
Supplementary Figure 1. Sabates-Bellver et al. * Supplementary Figure 1. Unsupervised hierarchical clustering of normal colonic mucosa (32 samples, green branches), colorectal adenomas (32 samples, red branches), and advanced colorectal carcinomas (25 samples, black branches) based on transcriptome analysis with Affymetrix U133 Plus 2.0 arrays. Data were analyzed as described in Methods. The 32,813 probes plotted on the y-axis (i.e., those expressed in at least one of the three tissue groups) are color-coded to indicate their expression levels relative to the median level for the gene across the entire sample set (blue: lower; red: higher). As expected, the tissue sample collected from patient NM’s polyp (*) clustered with the adenomas. This sample was clearly adenomatous although the polyp itself was classified as a T1 tumor (Table 1). Supplementary Figure 2; Sabates-Bellver et al. A B mitosis humoral defense mechanism CDC25B CCR2 CDC2 CCL14 CCNF C7 RAN CCL14 CDCA5 TNFRSF17 MAD2L1 CLU MAD2L1 CR2 CCNB1 C1S PRMT5 TNFRSF7 BUB1B SERPING1 PBK CD53 TPX2 KLRK1 BUB1 LY9 CDC2 CSF1R MAD2L2 KLRC4 CDC20 CLEC2B CDC6 C1QC UBE2C C1QA NUSAP1 C1QB AURKA HHEX BUB1 C1S CDC2 KLRC1 CCNA2 KLRD1 HCAP-G ITGB2 PTTG1 SPINK5 SMC2L1 HHEX PLK1 CD163 CCNA2 CCR2 KNTC2 MAG NEK2 CD163 KIF2C C3 BRRN1 XCL1 CCNB2 XCL2 NOLC1 ITGB2 KIF23 KLRB1 HCAP-G SLAMF1 KIF2C KLRD1 RCC1 LY9 AURKA CLEC2D CCNB1 CD1C CDC25C CD53 KIF11 CR1 ASPM CD74 CDC6 IGSF2 NEK2 CLEC2D KIFC1 CLU CDC25C CLU CENPE CCL18 NUSAP1 KLRF1 CDC25A CCL18 CCNF CR1 CDC25A CD40 CDC2 CCR7 adenomas normals CCR6 adenomas normals C D inflammatory response inflammatory response IL8 CCR2 ALOX5 NR3C1 IL8 NR3C1 MIF NR3C1 CXCL11 CXCL12 CXCL3 NR3C1 RIPK2 NR3C1 TLR4 AOC3 CXCL1 CD14 CXCL11 TLR7 CXCL2 STAB1 PF4 CXCL13 TLR4 CCL23 TLR4 HDAC9 CCL20 CCL21 TLR4 CCL5 CCL24 CCL5 S100A8 LY86 ALOX5 ITGAL S100A9 CCL23 REG3A CCL8 ANXA1 STAB1 SERPINA3 CCL5 IL1B CXCL12 IL1B CCL19 CCL26 BDKRB1 adenomas normals IL1R1 SIGLEC1 ADORA3 CCL2 PREX1 TLR3 Supplementary Figure 2.