21.9 Reduction of Carboxylic Acid Derivatives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Amide, and Paratoluenesulfonamide on the Amide of Silver,On the Imides
68 CHEMISTRY: E. C. FRANKLIN METALLIC SALTS OF AMMONO ACIDS By Edward C. Franklin DEPARTMENT OF CHEMISTRY, STANFORD UNIVERSITY Presented to the Academy, January 9. 1915 The Action of Liquid-Ammonia Solutions of Ammono Acids on Metallic Amides, Imides, and Nitrides. The acid amides and imides, and the metallic derivatives of the acid amides and imides are the acds, bases, and salts respectively of an ammonia system of acids, bases, and salts.1 Guided by the relationships implied in the above statement Franklin and Stafford were able to prepare potassium derivatives of a considerable number of acid amides by the action of potassium amide on certain acid amides in solution in liquid ammonia. That is to say, an ammono base, potassium amide, was found to react with ammono acids in liquid ammonia to form ammono salts just as the aquo base, potassium hydrox- ide, acts upon aquo acids in water solution to form aquo salts. Choos- ing, for example, benzamide and benzoic acid as representative acids of the two systems, the analogous reactions taking place respectively in liquid ammonia and water are represented by the equations: CH6CONH2+KNH2 = C6H5CONHK + NHs. CseHCONH2 + 2KNH2 = CIHsCONK2 + 2NH3. CH6tCOOH + KOH = CIH6COOK + H2O. The ammono acid, since it is dibasic, reacts with either one or two molecules of potassium amide to form an acid and a neutral salt. Having thus demonstrated the possibility of preparing ammono salts of potassium by the interaction of potassium amide and acid amides in liquid ammonia solution, it was further found that ammono salts of the heavy metals may be prepared by the action of liquid ammonia solutions of ammono acids on insoluble metallic amides, imides, and nitrides-that is, by reactions which are analogous to the formation of aquo salts in water by the action of potassium hydroxide on insoluble metallic hydroxides and oxides. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -
![United States Patent [191 [11] Patent Number: 5,070,175 Tsumura Et Al](https://docslib.b-cdn.net/cover/3510/united-states-patent-191-11-patent-number-5-070-175-tsumura-et-al-583510.webp)
United States Patent [191 [11] Patent Number: 5,070,175 Tsumura Et Al
United States Patent [191 [11] Patent Number: 5,070,175 Tsumura et al. [45] Date of Patent: Dec. 3, 1991 [54] METHOD FOR THE PREPARATION OF AN Primary Examiner-Morton Foelak ORGANOPOLYSILOXANE CONTAINING Attorney, Agent, or Firm-Millen, White & Zelano TETRAFUNCI'IONAL SILOXANE UNITS [57] ABSTRACT [75] Inventors: Hiroshi Tsumura; Kiyoyuki Mutoh, An ef?cient and economically advantageous method is both of Gunma; Kazushi Satoh, proposed for the preparation of an organopolysiloxane Tokyo; Ken-ichi Isobe, Gunma, all of comprising tetrafunctional siloxane units, i.e. Q units, Japan and, typically, monofunctional siloxy units, i.e. M units, [73] Assignee: Shin-Etsu Chemical Co., Ltd., Tokyo, and useful as a reinforcing agent in silicone rubbers. The Japan method comprises the steps of: mixing the reactants for providing the Q and M units, such as ethyl orthosilicate [21] Appl. No.;. 706,148 and trimethyl methoxy silane, in a desired molar ratio; [22] Filed: May 28, 1991 and heating the mixture at a temperature higher by at least 10° C. than the boiling point of the mixture under [30] Foreign Application Priority Data normal pressure in a closed vessel in the presence of May 29, 1990 [JP] Japan ............ .Q .................. .. 2-l39ll9 water and a catalyst such as a sulfonic acid group-con taining compound. In addition to the greatly shortened [51] Int. 01.5 ............................................ .. C08G 77/06 reaction time and remarkably decreased contents of [52] U.S. c1. ...................................... .. 528/12; 528/10; residual alkoxy groups and gelled matter in the product, 528/21; 528/23; 528/34; 528/36 the method is advantageous also in respect of the ab [58] Field of Search .................... -

NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION by JAMES ERVIN SCHAMMERHORN III Associates of Science Murray
NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC AROMATIC SUBSTITUTION By JAMES ERVIN SCHAMMERHORN III Associates of Science Murray State College Tishomingo, Oklahoma 2003 Bachelor of Science in Chemistry Oklahoma State University Stillwater, Oklahoma 2006 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY May, 2011 1 NEW TANDEM REACTIONS INVOLVING NUCLEOPHILIC ARMOMATIC SUBSTITUTION Thesis Approved: Dr. Richard Bunce Thesis Adviser Dr. K. Darrell Berlin Dr. Ziad El-Rassi Dr. Nicholas Materer Dr. Andrew Mort Dr. Mark E. Payton Dean of the Graduate College ii ACKNOWLEDGMENTS I would like to express my sincere gratitude to Dr. R. A. Bunce, my research advisor, my graduate committee chairman and my friend. I have learned many valuable lessons working alongside him in his research laboratory. His ability to teach both organic theory and laboratory techniques have been instrumental in my research at Oklahoma State University. I am immensely thankful for his support, guidance and patience to me during the course of this research. His unique sense of humor always makes me laugh and makes it a joy to come to lab. I would also like to thank my committee members, Drs. K. D. Berlin, Z. El Rassi, N. F. Materer, and A. J. Mort their acceptance to be on my committee. Their insights into research and graduate life have been invaluable during my graduate career. I am especially grateful to Dr. Berlin for sharing his wealth of organic chemistry knowledge with me during the course of my research. Also, to Dr. -

Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction
Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction Mary A. Yohannan* and Arthur Berrier U.S. Department of Justice Drug Enforcement Administration Special Testing and Research Laboratory 22624 Dulles Summit Court Dulles, VA 20166 [email: mary.a.yohannan -at- usdoj.gov] ABSTRACT: The formation of imine by-products from phenethylamines, amphetamines, and tryptamines upon an acetone extraction is presented. These imine by-products were characterized using GC/MSD and exhibited preferential cleavage at the α-carbon of the alkyl chain. Further characterization of the imine by-products of phenethylamine and tryptamine was done using IR and NMR. KEYWORDS: phenethylamine, tryptamine, imine, acetone, schiff base, drug chemistry, forensic chemistry In most forensic laboratories, the solvents used to extract at the α-carbon on the alkyl chain. In addition to GC/MS, the drugs are chosen based upon their solubility properties and their imines formed from phenethylamine base and tryptamine base ability to not interact with the drug. In fact, there are very few were characterized by Fourier transform-infrared spectroscopy publications where a solvent used to extract a drug reacts with (FTIR) and nuclear magnetic resonance (NMR) spectroscopy. the drug and forms by-products [1-3]. This laboratory recently discovered that an additional Experimental component was formed when acetone was used to extract a Solvents, Chemicals, and Materials sample containing a known tryptamine. Analysis by gas Acetone was ACS/HPLC grade from Burdick and Jackson chromatography/mass spectroscopy (GC/MS) of the acetone Laboratories (Muskegon, MI). Phenethylamine base and extract yielded an extra peak in the total ion chromatogram that tryptamine base were obtained from Sigma-Aldrich Chemicals was approximately half the abundance of the known tryptamine (Milwaukee, WI). -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

The Carcinogenicity of the O-Methoxy Derivatives of N-2-Fluorenylacetamide and of Related Compounds in the Rat
[CANCER RESEARCH 28, 234-244, February 1968] The Carcinogenicity of the o-Methoxy Derivatives of N-2-Fluorenylacetamide and of Related Compounds in the Rat H. R. Gutmann, S. B. Galitski, and W. A. Foley Laboratory ]or Cancer Research, Veterans Administration Hospital, and Department o] Biochemistry, University o] Minnesota, Minneapolis, Minnesota 55417 SUMMARY acetamide by the sequential reactions of deacetylation and oxidation. In order to test the idea that the lack of carcinogenicity of the o-amidofluorenols, N- (1-hydroxy-2-fluorenyl) acetamide and INTRODUCTION N-(3-hydroxy-2-fluorenyl)acetamide, is due to the hydrophilic phenolic hydroxyl group, the methylated derivatives, N-(1- Several model studies from this laboratory have shown that methoxy-2-fluorenyl)acetamide and N-(3-methoxy-2-fluorenyl) the o-quinone imines, 2-imino-l,2-fluorenoquinone and 2-imino- acetamide as well as the hydrochlorides of 1-methoxy-2-fluo- 2,3-fluorenoquinone, which are derived from the carcinogen renamine and 3-methoxy-2-fiuorenamine, were prepared, and N-2-fluorenylacetamide by the sequential enzymatic reactions their carcinogenicity was evaluated in the rat. N-(1-Methoxy- of hydroxylation, deacetylation, and oxidation (6, 10, 12, 27, 2-fluorenyl)acetamide and 1-methoxy-2-fluorenamine hydro- 34, 35), form stable adducts with a variety of proteins (17, 18). chloride, when administered orally to male rats for 5 months, However, the relevance of the binding of these o-quinone imines gave a tumor incidence of 27 and 50%, respectively. Approxi- to chemical carcinogenesis has remained obscure largely be- mately one-half of the lesions produced by either com- cause the o-amidofluorenols, 1-OH-AAF 2 and 3-OH-AAF pound were adenocarcinomas of the small intestine. -
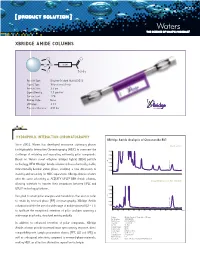
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj.