Carbon-Halogen Bond Activation by a Structurally Constrained
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Dielectric Constant Chart
Dielectric Constants of Common Materials DIELECTRIC MATERIALS DEG. F CONSTANT ABS RESIN, LUMP 2.4-4.1 ABS RESIN, PELLET 1.5-2.5 ACENAPHTHENE 70 3 ACETAL 70 3.6 ACETAL BROMIDE 16.5 ACETAL DOXIME 68 3.4 ACETALDEHYDE 41 21.8 ACETAMIDE 68 4 ACETAMIDE 180 59 ACETAMIDE 41 ACETANILIDE 71 2.9 ACETIC ACID 68 6.2 ACETIC ACID (36 DEGREES F) 36 4.1 ACETIC ANHYDRIDE 66 21 ACETONE 77 20.7 ACETONE 127 17.7 ACETONE 32 1.0159 ACETONITRILE 70 37.5 ACETOPHENONE 75 17.3 ACETOXIME 24 3 ACETYL ACETONE 68 23.1 ACETYL BROMIDE 68 16.5 ACETYL CHLORIDE 68 15.8 ACETYLE ACETONE 68 25 ACETYLENE 32 1.0217 ACETYLMETHYL HEXYL KETONE 66 27.9 ACRYLIC RESIN 2.7 - 4.5 ACTEAL 21 3.6 ACTETAMIDE 4 AIR 1 AIR (DRY) 68 1.000536 ALCOHOL, INDUSTRIAL 16-31 ALKYD RESIN 3.5-5 ALLYL ALCOHOL 58 22 ALLYL BROMIDE 66 7 ALLYL CHLORIDE 68 8.2 ALLYL IODIDE 66 6.1 ALLYL ISOTHIOCYANATE 64 17.2 ALLYL RESIN (CAST) 3.6 - 4.5 ALUMINA 9.3-11.5 ALUMINA 4.5 ALUMINA CHINA 3.1-3.9 ALUMINUM BROMIDE 212 3.4 ALUMINUM FLUORIDE 2.2 ALUMINUM HYDROXIDE 2.2 ALUMINUM OLEATE 68 2.4 1 Dielectric Constants of Common Materials DIELECTRIC MATERIALS DEG. F CONSTANT ALUMINUM PHOSPHATE 6 ALUMINUM POWDER 1.6-1.8 AMBER 2.8-2.9 AMINOALKYD RESIN 3.9-4.2 AMMONIA -74 25 AMMONIA -30 22 AMMONIA 40 18.9 AMMONIA 69 16.5 AMMONIA (GAS?) 32 1.0072 AMMONIUM BROMIDE 7.2 AMMONIUM CHLORIDE 7 AMYL ACETATE 68 5 AMYL ALCOHOL -180 35.5 AMYL ALCOHOL 68 15.8 AMYL ALCOHOL 140 11.2 AMYL BENZOATE 68 5.1 AMYL BROMIDE 50 6.3 AMYL CHLORIDE 52 6.6 AMYL ETHER 60 3.1 AMYL FORMATE 66 5.7 AMYL IODIDE 62 6.9 AMYL NITRATE 62 9.1 AMYL THIOCYANATE 68 17.4 AMYLAMINE 72 4.6 AMYLENE 70 2 AMYLENE BROMIDE 58 5.6 AMYLENETETRARARBOXYLATE 66 4.4 AMYLMERCAPTAN 68 4.7 ANILINE 32 7.8 ANILINE 68 7.3 ANILINE 212 5.5 ANILINE FORMALDEHYDE RESIN 3.5 - 3.6 ANILINE RESIN 3.4-3.8 ANISALDEHYDE 68 15.8 ANISALDOXINE 145 9.2 ANISOLE 68 4.3 ANITMONY TRICHLORIDE 5.3 ANTIMONY PENTACHLORIDE 68 3.2 ANTIMONY TRIBROMIDE 212 20.9 ANTIMONY TRICHLORIDE 166 33 ANTIMONY TRICHLORIDE 5.3 ANTIMONY TRICODIDE 347 13.9 APATITE 7.4 2 Dielectric Constants of Common Materials DIELECTRIC MATERIALS DEG. -

Particularly Hazardous Substances (PHS)
Chemical Class Standard Operating Procedures Particularly Hazardous Substances (PHS) Print a copy and insert into your laboratory SOP binder. Department: Chemistry Date SOP was written: June 10, 2013 Date SOP was approved by PI/lab supervisor: Name: Richmond Sarpong Principal Investigator: Signature: ______________________________ Name: Rebecca Murphy Internal Lab Safety Coordinator or Lab Manager: Lab Phone: 510-643-2485 Office Phone: 510-642-6312 Name: Richmond Sarpong Emergency Contact: Phone Number: 626-644-2407 Latimer Hall: 834, 836, 837, 838, 839, 842, 847, 849, Location(s) covered by this SOP: 907 1. Purpose This SOP covers the precautions and safe handling procedures for the use of Particularly Hazardous Substances in the Sarpong group, which include the following chemicals and their uses: Chemical(s) Use(s) PHSs 1. Using Particularly Hazardous Substances as reagents. PHS solvents 2. Using Particularly Hazardous Substances as solvents in reactions, for extractions or cleaning glassware. PHS solvents 3. Using Particularly Hazardous Substances as solvents in column chromatography (CC) or thin layer chromatography (TLC). Sarpong inventory chemicals See below. covered by this SOP Rev. Date: June 10, 2013 1 Particularly Hazardous Substances Class SOP Sarpong Group inventory chemicals covered by this SOP (R)-(+)-2,3-Dihydroindole-2-carboxylic acid Chloroform (r)-styrene oxide cis-1,4-Dichloro-2-butene (S)-(-)-Indoline-2-carboxylic acid Cobalt chloride hexahydrate 1,1,3,3-Tetramethylurea Cobalt(II) acetylacetonate 1,2,4-Triazole Cyclopropylboronic -

Free Radical Reactions of Allylic and Propargylic Derivatives Yuh-Wern Wu Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1990 Free radical reactions of allylic and propargylic derivatives Yuh-Wern Wu Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Wu, Yuh-Wern, "Free radical reactions of allylic and propargylic derivatives " (1990). Retrospective Theses and Dissertations. 9473. https://lib.dr.iastate.edu/rtd/9473 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS The most advanced technology has been used to photograph and reproduce this manuscript from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -
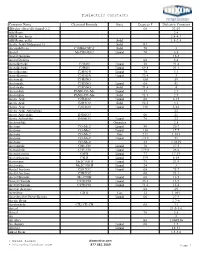
Dielectric Constants
Dielectric Constants Common Name Chemical Formula State Degrees F Dielectric Constant (Ethylenedioxy)diethanol-2,2` 68 23.69 ABS Resin 2.4 ABS Resin, lump 2.4-4.1 ABS Resin, pellet Solid 1.5-2.5 Acatic.Acid (36degrees F) Solid 4.1 Acenaphthene C10H6(CH2)2 Solid 70 3 Acetal MeCH(OEt)2 Liquid 70 3.6 Acetal Bromide 16.5 Acetal Doxime 68 3.4 Acetaldehyde C2H4O Liquid 50 21.8 Acetaldehyde C2H4O Liquid 69.8 21.1 Acetaldoxime C2H5ON Liquid 70 3.4 Acetaldoxime C2H5ON Liquid 73.4 3 Acetamide C3H5NO 180 59 Acetamide C3H5NO Liquid 68 41 Acetamide C3H5NO Solid 71.6 4 Acetanilide PhNH-CO-Me Liquid 71 2.9 Acetanilide PhNH-CO-Me Solid 71.6 2.9 Acetic Acid C2H4O2 Liquid 68 6.15 Acetic Acid C2H4O2 Solid 32.5 4.1 Acetic Acid C2H4O2 Liquid 158 6.62 Acetic Acid, Anhydrous 22 Acetic Anhydride H4H6O3 66 21 Acetic Anhydride H4H6O3 Liquid 70 22 Acetoanilide Granules 2.8 Acetone CO-Me2 Liquid 80 20.7 Acetone CO-Me2 Liquid 130 17.7 Acetone CO-Me2 Gas 212 1.015 Acetone CO-Me2 Liquid -112 31 Acetone CO-Me2 32 1.0159 Acetonitrile CH3-CN Liquid 70 37.5 Acetonitrile CH3-CN Liquid 179.6 26.6 Acetophenone C8H8 Liquid 77 17.39 Acetophenone C8H8 Liquid 394 8.64 Acetoxime Me2C:NOH Liquid 75 23.9 Acetoxime Me2C:NOH Liquid 24 3 Acetyl Acetone C5H2O2 Liquid 68 25.7 Acetyl Acetone C5H2O2 68 23.1 Acetyl Bromide Me-COBr Liquid 68 16.5 Acetyl Chloride C2H3ClO Liquid 35.6 16.9 Acetyl Chloride C2H3ClO Liquid 71.6 15.8 Acetyle Acetone 68 25 Acetylene C2H2 Gas 32 1.001 Acetylmethyl Hexyl Ketone Liquid 66 27.9 Acrylic Resin 2.7-6 Acrylonitrile CH2:CH-CN 68 33 Acteal 21.0-3.6 Acteal -

UPS Chemical Table
UPS Chemical Table - 49 CFR Version (Ground and Air Packages) BASIC DESCRIPTION FOR GROUND AND AIR GROUND AND AIR HAZARDOUS MATERIALS SHIPMENTS GROUND SHIPMENTS AIR SHIPMENTS SHIPMENTS Symbols: "‡" Requires a Technical Name / "*" For Ltd Qty see 49 CFR Part 173.*** / "**" See 49 CFR 173.27 for inner receptacle requirements / "▲" Accepted as CAO if amount is listed / "♦" PG = "II" or blank / "♦♦" PG = "III" or blank / "#" Sub-risk may be omitted from shipping papers if 49 CFR 172.400a(c) criteria met. (ref 172.202(a)(3)) CARGO HAZARD DOT PASSENGER AIRCRAFT DOT CLASS OR EXEMPTION, GROUND LTD QTY AIRCRAFT MAX NET EXEMPTION, HAZARDOUS MATERIALS DESCRIPTIONS DIVISION I.D. NUMBER LABEL(S) REQUIRED OR SPECIAL SERVICE TO LABEL(S) REQUIRED OR MAX NET MAX NET PER SPECIAL NON-BULK AND PROPER SHIPPING NAME (Subsidiary if (ALSO MARK PACKING EXEMPTION, SPECIAL PERMIT OR PERMIT CANADA EXEMPTION, SPECIAL PERMIT OR PER PER PACKAGE PERMIT SPECIAL EXCEPTIONS PACKAGING (ALSO MARK ON PACKAGE) applicable) ON PACKAGE) GROUP EXCEPTION OR 173.13 PERMITTED EXCEPTION PACKAGE** PACKAGE** ▲ OR 173.13 PROVISIONS §173.*** §173.*** (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flamable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flamable gas) Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic -

Alcohols, Phenols and Ethers Alcohols Preparation of Alcohols: by Hydrolysis of Haloalkanes : R-X + Aq
Alcohols, Phenols and Ethers Alcohols Preparation of Alcohols: By hydrolysis of haloalkanes : R-X + aq. KOH →ROH +KX By reduction of Carbonyl compounds By the action of Grignard’s Reagent on aldehydes, ketones and esters By Aliphatic Primary Amines: RCH2NH2 + HNO2 → RCH2OH + N2 + H2O Hydration of alkenes: Oxymercuration-demercuration: Hydroboration-oxidation: Hydroxylation of alkenes: Physical Properties of Alcohol: Lower alcohols are liquid at room temperature while higher ones are solid. High boiling point due to presence of intermolecular hydrogen bonding. Order of Boiling Point: primary > secondary > tertiary Solubility in water decreases with increase in molecular mass due to decrease in extent of intermolecular hydrogen bonding. Chemical Properties of Alcohol: + – Alcohol's reaction with metal: ROH + Na→2RO Na + H2 Formation of Halides: o 3ROH + P+I2→3RI + H3PO3 o ROH + SOCl2/PCl3/PCl5→ RCl o ROH+HX→ RX o ROH+ NaBr,H2SO4→R-Br o ROH+ Zn+HCl→R-Cl o R2C-OH alcohol + HCl→ R2CCl Reaction with HNO3: R-OH + HO-NO2→ R-O-NO2 Reaction with carboxylic acid (Esterification) : R-OH +R’-COOH +H+↔ R’-COOR Reaction with Grignard reagent: R'OH + RMgX → RH + R'OMgX Reduction of alcohol : ROH + 2HI + Red P→ RH +I2+H2O Dehydration of Alcohol: Dehydration of alcohols takes place in acidic medium. Intra- molecular dehydration leads to the formation of alkene while inter molecular dehydration which forms ether. Ease of dehydration: 3° > 2° > 1 Satyzeff’s Rule : Elimination through b carbon containing minimum b hydrogen Oxidation of Alcohol: RCH2-OH + [O] → RCHO → RCOOH RCH2-OH + [O] +PCC → RCHO Haloform Reaction: Compound containing CH3CO- group (or compound on oxidation gives CH3CO – group) which is attached with a C or H, in presence of halogen and mild alkali gives haloform.CH3-CH2-COCH2-CH3, CH3-CO-Cl, CH3COOH will not respond to haloform reaction wile CH3CH2OH will respond to haloform Reaction. -

Hazardous Chemicals Handbook
Hazardous Chemicals Handbook Hazardous Chemicals Handbook Second edition Phillip Carson PhD MSc AMCT CChem FRSC FIOSH Head of Science Support Services, Unilever Research Laboratory, Port Sunlight, UK Clive Mumford BSc PhD DSc CEng MIChemE Consultant Chemical Engineer Oxford Amsterdam Boston London New York Paris San Diego San Francisco Singapore Sydney Tokyo Butterworth-Heinemann An imprint of Elsevier Science Linacre House, Jordan Hill, Oxford OX2 8DP 225 Wildwood Avenue, Woburn, MA 01801-2041 First published 1994 Second edition 2002 Copyright © 1994, 2002, Phillip Carson, Clive Mumford. All rights reserved The right of Phillip Carson and Clive Mumford to be identified as the authors of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988 No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London, England W1T 4LP. Applications for the copyright holder’s written permission to reproduce any part of this publication should be addressed to the publishers British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Cataloguing -

Hazardous Materials Shipping Guide
Hazardous Materials Shipping Guide FedEx Ground Package Systems Inc. is committed to the safe transportation of hazardous materials. It is very important that each person engaged in the transportation of hazardous materials become thoroughly familiar with the Title 49CFR (Code of Federal Regulations). This guide is intended only to assist you in your preparation of hazardous materials shipped via FedEx Ground Package Systems Inc. It is the shipper’s responsibility to ensure each hazardous material package is in compliance with applicable Department of Transportation (D.O.T.) regulations and FedEx Ground Package Systems Inc. requirements. Failure to comply with these regulations and requirements may subject the shipper and carrier to fines and penalties. Due to the changing nature of D.O.T. regulations and other information, it is impossible to guarantee absolute accuracy of the material contained in this guide. FedEx Ground Package Systems Inc., therefore, cannot assume any responsibility for omissions, errors, misprinting, or ambiguity contained within this guide and shall not be held liable in any degree for any loss or injury caused by such omission or error presented in this publication. The FedEx Ground Hazardous Materials Shipping Guide is intended to simplify Title 49 CFR. FedEx Ground Package Systems Inc. reserves the right to be more restrictive than the federal regulations (49 CFR). Customers should be thoroughly familiar with the applicable sections of this guide when shipping hazardous materials via FedEx Ground Package Systems -

SEBASTIANO DE LUCA Alkenes, Glycerin, and Derivatives, Plant Principles
Revista CENIC. Ciencias Químicas ISSN: 1015-8553 ISSN: 2221-2442 [email protected] Centro Nacional de Investigaciones Científicas Cuba SEBASTIANO DE LUCA Alkenes, glycerin, and derivatives, plant principles Wisniak, Jaime SEBASTIANO DE LUCA Alkenes, glycerin, and derivatives, plant principles Revista CENIC. Ciencias Químicas, vol. 50, no. 1, 2019 Centro Nacional de Investigaciones Científicas, Cuba Available in: https://www.redalyc.org/articulo.oa?id=181662291005 PDF generated from XML JATS4R by Redalyc Project academic non-profit, developed under the open access initiative Jaime Wisniak. SEBASTIANO DE LUCA Alkenes, glycerin, and derivatives, plant principles Reseñas SEBASTIANO DE LUCA Alkenes, glycerin, and derivatives, plant principles SEBASTIANO DE LUCA Alquenos, glicerina y derivados, principios vegetales. Jaime Wisniak * Redalyc: https://www.redalyc.org/articulo.oa? Ben-Gurion University of the Negev, Israel id=181662291005 [email protected] Received: 31 July 2019 Accepted: 20 September 2019 Abstract: Sebastiano de Luca (1820-1880) was a multifaceted Italian-French chemist who carried on research on a wide variety of subjects, among them, the synthesis of numerous organic derivatives of iodine, the action of halogens upon glycerin, the separation of active vegetable principles, analytic, inorganic, physiological and atmospheric chemistry, formation of sugar in the liver and of oil in olives, as well as the analysis of the products derived from volcanic activity and of materials found in the ruins of Pompeii. He synthesized for the first time allyl iodide and pure propylene by the reaction between glycerin and phosphorus triiodide, and a large number of allyl derivatives (i.e. allyl ethyl, allyl phenyl allyl halides, allyl alcohol), and determined their physical and chemical properties.