Characterizing Alternative Splicing and Long Non
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of Copenhagen, Copenhagen, Denmark, Citation: Pereira J, Johnson WE, O’Brien SJ, 8
Evolutionary genomics and adaptive evolution of the hedgehog gene family (Shh, Ihh and Dhh) in vertebrates Pereira, Joana; Johnson, Warren E.; O'Brien, Stephen J.; Jarvis, Erich D; Zhang, Guojie; Gilbert, M. Thomas P.; Vasconcelos, Vitor; Antunes, Agostinho Published in: PloS one DOI: 10.1371/journal.pone.0074132 Publication date: 2014 Document version Publisher's PDF, also known as Version of record Document license: CC BY Citation for published version (APA): Pereira, J., Johnson, W. E., O'Brien, S. J., Jarvis, E. D., Zhang, G., Gilbert, M. T. P., Vasconcelos, V., & Antunes, A. (2014). Evolutionary genomics and adaptive evolution of the hedgehog gene family (Shh, Ihh and Dhh) in vertebrates. PloS one, 9(12), [e74132]. https://doi.org/10.1371/journal.pone.0074132 Download date: 24. sep.. 2021 RESEARCH ARTICLE Evolutionary Genomics and Adaptive Evolution of the Hedgehog Gene Family (Shh, Ihh and Dhh) in Vertebrates Joana Pereira1¤, Warren E. Johnson2, Stephen J. O’Brien3,4, Erich D. Jarvis5, Guojie Zhang6, M. Thomas P. Gilbert7, Vitor Vasconcelos1,8, Agostinho Antunes1,8* 1. CIIMAR/CIMAR, Interdisciplinary Centre of Marine and Environmental Research, University of Porto, Porto, Portugal, 2. Smithsonian Conservation Biology Institute, National Zoological Park, Front Royal, Virginia, United States of America, 3. Theodosius Dobzhansky Center for Genome Bioinformatics, St. Petersburg State University, St. Petersburg, Russia, 4. Oceanographic Center, N. Ocean Drive, Nova Southeastern University, Ft. Lauderdale, Florida, United States of America, 5. Howard Hughes Medical Institute, Department of Neurobiology, Duke University Medical Center, Durham, North Carolina, United States of America, 6. BGI-Shenzhen, Beishan Industrial Zoon, Yantian District, Shenzhen, China, 7. -

Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 Are Associated with Hearing Loss in Human and Mouse
RESEARCH ARTICLE Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in human and mouse Rizwan Yousaf1, Chunfang Gu2, Zubair M. Ahmed3, Shaheen N. Khan4, Thomas B. Friedman5, Sheikh Riazuddin6, Stephen B. Shears2, Saima Riazuddin1* a1111111111 1 Laboratory of Molecular Genetics, Department of Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United States of America, 2 Signal Transduction Laboratory, a1111111111 National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, a1111111111 NC, United States of America, 3 Laboratory of Neurogenetics and Regenerative Medicine, Department of a1111111111 Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United a1111111111 States of America, 4 National Center for Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan, 5 Section on Human Genetics, Laboratory of Molecular Genetics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD, United States of America, 6 Shaheed Zulfiqar Ali Bhutto Medical University, Pakistan Institute of Medical Sciences, Islamabad, Pakistan * [email protected] OPEN ACCESS Citation: Yousaf R, Gu C, Ahmed ZM, Khan SN, Friedman TB, Riazuddin S, et al. (2018) Mutations Abstract in Diphosphoinositol-Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in Autosomal recessive nonsyndromic hearing loss is a genetically heterogeneous disorder. human and mouse. PLoS Genet 14(3): e1007297. https://doi.org/10.1371/journal.pgen.1007297 Here, we report a severe-to-profound sensorineural hearing loss locus, DFNB100 on chro- mosome 5q13.2-q23.2. Exome enrichment followed by massive parallel sequencing Editor: Karen B. Avraham, Tel Aviv University, ISRAEL revealed a c.2510G>A transition variant in PPIP5K2 that segregated with DFNB100-associ- ated hearing loss in two large apparently unrelated Pakistani families. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

FUSIP1 Polyclonal Antibody Catalog Number PA5-41929 Product Data Sheet
Lot Number: A9C701N Website: thermofisher.com Customer Service (US): 1 800 955 6288 ext. 1 Technical Support (US): 1 800 955 6288 ext. 441 thermofisher.com/contactus FUSIP1 Polyclonal Antibody Catalog Number PA5-41929 Product Data Sheet Details Species Reactivity Size 100 µl Tested species reactivity Equine, Guinea Pig, Human, Mouse, Host / Isotype Rabbit IgG Rabbit, Rat Class Polyclonal Tested Applications Dilution * Type Antibody Immunohistochemistry (Paraffin) 4-8 µg/mL (IHC (P)) Immunogen Synthetic peptide directed towards the C-terminal of human FUSIP1 Western Blot (WB) 0.2-1 µg/mL Conjugate Unconjugated * Suggested working dilutions are given as a guide only. It is recommended that the user titrate the product for use in their own experiment using appropriate negative and positive controls. Form Liquid Concentration 0.5mg/mL Purification Affinity Chromatography Storage Buffer PBS with 2% sucrose Contains 0.09% sodium azide Storage Conditions -20° C, Avoid Freeze/Thaw Cycles Product Specific Information Peptide sequence: TDSKTHYKSG SRYEKESRKK EPPRSKSQSR SQSRSRSKSR SRSWTSPKSS Sequence homology: Guinea Pig: 100%; Horse: 100%; Human: 100%; Mouse: 100%; Rabbit: 100%; Rat: 100% Background/Target Information FUSIP1 is a member of the serine-arginine (SR) family of proteins, which is involved in constitutive and regulated RNA splicing. Members of this family are characterized by N-terminal RNP1 and RNP2 motifs, which are required for binding to RNA, and multiple C-terminal SR/RS repeats, which are important in mediating association with other cellular proteins. This protein can influence splice site selection of adenovirus E1A pre-mRNA. It interacts with the oncoprotein TLS, and abrogates the influence of TLS on E1A pre-mRNA splicing.This gene product is a member of the serine-arginine (SR) family of proteins, which is involved in constitutive and regulated RNA splicing. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

FUSIP1 (SRSF10) (NM 006625) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC221759 FUSIP1 (SRSF10) (NM_006625) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: FUSIP1 (SRSF10) (NM_006625) Human Tagged ORF Clone Tag: Myc-DDK Symbol: SRSF10 Synonyms: FUSIP1; FUSIP2; NSSR; PPP1R149; SFRS13; SFRS13A; SRp38; SRrp40; TASR; TASR1; TASR2 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC221759 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGTCCCGCTACCTGCGTCCCCCCAACACGTCTCTGTTCGTCAGGAACGTGGCCGACGACACCAGGTCTG AAGACTTGCGGCGTGAATTTGGTCGTTATGGTCCTATAGTTGATGTGTATGTTCCACTTGATTTCTACAC TCGCCGTCCAAGAGGATTTGCTTATGTTCAATTTGAGGATGTTCGTGATGCTGAAGACGCTTTACATAAT TTGGACAGAAAGTGGATTTGTGGACGGCAGATTGAAATACAGTTTGCCCAGGGGGATCGAAAGACACCAA ATCAGATGAAAGCCAAGGAAGGGAGGAATGTGTACAGTTCTTCACGCTATGATGATTATGACAGATACAG ACGTTCTAGAAGCCGAAGTTATGAAAGGAGGAGATCAAGAAGTCGGTCTTTTGATTACAACTATAGAAGA TCGTATAGTCCTAGAAACAGTAGACCGACTGGAAGACCACGGCGTAGCAGAAGCCATTCCGACAATGATA GACCAAACTGCAGCTGGAATACCCAGTACAGTTCTGCTTACTACACTTCAAGAAAGATC ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA Protein Sequence: >RC221759 protein sequence Red=Cloning site Green=Tags(s) MSRYLRPPNTSLFVRNVADDTRSEDLRREFGRYGPIVDVYVPLDFYTRRPRGFAYVQFEDVRDAEDALHN -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Evidence for Differential Alternative Splicing in Blood of Young Boys With
Stamova et al. Molecular Autism 2013, 4:30 http://www.molecularautism.com/content/4/1/30 RESEARCH Open Access Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders Boryana S Stamova1,2,5*, Yingfang Tian1,2,4, Christine W Nordahl1,3, Mark D Shen1,3, Sally Rogers1,3, David G Amaral1,3 and Frank R Sharp1,2 Abstract Background: Since RNA expression differences have been reported in autism spectrum disorder (ASD) for blood and brain, and differential alternative splicing (DAS) has been reported in ASD brains, we determined if there was DAS in blood mRNA of ASD subjects compared to typically developing (TD) controls, as well as in ASD subgroups related to cerebral volume. Methods: RNA from blood was processed on whole genome exon arrays for 2-4–year-old ASD and TD boys. An ANCOVA with age and batch as covariates was used to predict DAS for ALL ASD (n=30), ASD with normal total cerebral volumes (NTCV), and ASD with large total cerebral volumes (LTCV) compared to TD controls (n=20). Results: A total of 53 genes were predicted to have DAS for ALL ASD versus TD, 169 genes for ASD_NTCV versus TD, 1 gene for ASD_LTCV versus TD, and 27 genes for ASD_LTCV versus ASD_NTCV. These differences were significant at P <0.05 after false discovery rate corrections for multiple comparisons (FDR <5% false positives). A number of the genes predicted to have DAS in ASD are known to regulate DAS (SFPQ, SRPK1, SRSF11, SRSF2IP, FUS, LSM14A). In addition, a number of genes with predicted DAS are involved in pathways implicated in previous ASD studies, such as ROS monocyte/macrophage, Natural Killer Cell, mTOR, and NGF signaling. -

Genome‐Wide Association Studies of the Self‐Rating of Effects of Ethanol (SRE)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by eScholarship - University of California UC San Diego UC San Diego Previously Published Works Title Genome-wide association studies of the self-rating of effects of ethanol (SRE). Permalink https://escholarship.org/uc/item/94p1n78c Journal Addiction biology, 25(2) ISSN 1355-6215 Authors Lai, Dongbing Wetherill, Leah Kapoor, Manav et al. Publication Date 2020-03-01 DOI 10.1111/adb.12800 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Received: 18 December 2018 Revised: 6 May 2019 Accepted: 27 May 2019 DOI: 10.1111/adb.12800 ORIGINAL ARTICLE Genome‐wide association studies of the self‐rating of effects of ethanol (SRE) Dongbing Lai1 | Leah Wetherill1 | Manav Kapoor2 | Emma C. Johnson3 | Melanie Schwandt4 | Vijay A. Ramchandani5 | David Goldman4 | Geoff Joslyn6 | Xi Rao1 | Yunlong Liu1 | Sean Farris7 | R. Dayne Mayfield7 | Danielle Dick8 | Victor Hesselbrock9 | John Kramer10 | Vivia V. McCutcheon3 | John Nurnberger1,11 | Jay Tischfield12 | Alison Goate2 | Howard J. Edenberg1,13 | Bernice Porjesz14 | Arpana Agrawal3 | Tatiana Foroud1 | Marc Schuckit15 1 Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, Indiana 2 Department of Neuroscience, Icahn School of Medicine at Mt. Sinai, New York, New York 3 Department of Psychiatry, Washington University School of Medicine, St. Louis, Missouri 4 Office of the Clinical Director, National Institute on Alcohol Abuse -

Supplementary Materials For
www.sciencemag.org/cgi/content/full/science.1230422/DC1 Supplementary Materials for Genomic Diversity and Evolution of the Head Crest in the Rock Pigeon Michael D. Shapiro,* Zev Kronenberg, Cai Li, Eric T. Domyan, Hailin Pan, Michael Campbell, Hao Tan, Chad D. Huff, Haofu Hu, Anna I. Vickrey, Sandra C. A. Nielsen, Sydney A. Stringham, Hao Hu, Eske Willerslev, M. Thomas P. Gilbert, Mark Yandell, Guojie Zhang, Jun Wang* *To whom correspondence should be addressed. E-mail: [email protected] (M.D.S.); [email protected] (J.W.) Published 31 January 2013 on Science Express DOI: 10.1126/science.1230422 This PDF file includes: Materials and Methods Supplementary Text Figs. S1 to S27 Tables S1 to S28 References (26–72) Materials and Methods Genome assembly The DNA sample for sequencing of the reference genome was extracted from blood obtained from a single, male Danish Tumbler, bred by Anders and Hans Ove Christiansen (Danmarks Racedueforeninger, Næstved, Denmark). This breed was chosen because it is an old breed that is believed to have changed little in recent history. Seven paired-end sequencing libraries were constructed, with insert sizes of 170 bp, 500 bp, 800 bp, 2 kb, 5 kb, 10 kb and 20 kb. The libraries were sequenced using Illumina HiSeq2000 platform, yielding a total of 127.17 Gb raw data (Table S1). The raw sequences were filtered for low quality, adapter sequence, paired-end read overlap, and PCR duplicates. We also performed an error correction step on the raw reads before assembling. Filtering and error correction resulted in 81.57 Gb of clean data for genome assembly with the genome with SOAPdenovo (26). -

RNF2 Antibody Order 021-34695924 [email protected] Support 400-6123-828 50Ul [email protected] 100 Ul √ √ Web
TD7403 RNF2 Antibody Order 021-34695924 [email protected] Support 400-6123-828 50ul [email protected] 100 uL √ √ Web www.ab-mart.com.cn Description: E3 ubiquitin-protein ligase that mediates monoubiquitination of 'Lys-119' of histone H2A (H2AK119Ub), thereby playing a central role in histone code and gene regulation. H2AK119Ub gives a specific tag for epigenetic transcriptional repression and participates in X chromosome inactivation of female mammals. May be involved in the initiation of both imprinted and random X inactivation (By similarity). Essential component of a Polycomb group (PcG) multiprotein PRC1-like complex, a complex class required to maintain the transcriptionally repressive state of many genes, including Hox genes, throughout development. PcG PRC1 complex acts via chromatin remodeling and modification of histones, rendering chromatin heritably changed in its expressibility. E3 ubiquitin-protein ligase activity is enhanced by BMI1/PCGF4. Acts as the main E3 ubiquitin ligase on histone H2A of the PRC1 complex, while RING1 may rather act as a modulator of RNF2/RING2 activity (Probable). Association with the chromosomal DNA is cell-cycle dependent. In resting B- and T-lymphocytes, interaction with AURKB leads to block its activity, thereby maintaining transcription in resting lymphocytes (By similarity). Uniprot:Q99496 Alternative Names: BAP 1; BAP1; DING; DinG protein; E3 ubiquitin protein ligase RING 2; E3 ubiquitin protein ligase RING2; E3 ubiquitin-protein ligase RING2; HIP2 interacting protein 3; HIP2- interacting protein 3; HIPI 3; HIPI3; Huntingtin interacting protein 2 interacting protein 3; Huntingtin-interacting protein 2-interacting protein 3; OTTHUMP00000060668; Polycomb M33 interacting protein Ring 1B; Polycomb M33 interacting protein Ring1B; Protein DinG; RING 1B; RING 2; RING finger protein 1B; RING finger protein 2; RING finger protein BAP 1; RING finger protein BAP-1; RING finger protein BAP1; RING1b; RING2_HUMAN; RNF 2; Rnf2; Specificity: RNF2 Antibody detects endogenous levels of total RNF2. -
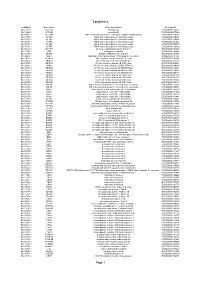
Epigenetics Page 1
Epigenetics esiRNA ID Gene Name Gene Description Ensembl ID HU-13237-1 ACTL6A actin-like 6A ENSG00000136518 HU-13925-1 ACTL6B actin-like 6B ENSG00000077080 HU-14457-1 ACTR1A ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) ENSG00000138107 HU-10579-1 ACTR2 ARP2 actin-related protein 2 homolog (yeast) ENSG00000138071 HU-10837-1 ACTR3 ARP3 actin-related protein 3 homolog (yeast) ENSG00000115091 HU-09776-1 ACTR5 ARP5 actin-related protein 5 homolog (yeast) ENSG00000101442 HU-00773-1 ACTR6 ARP6 actin-related protein 6 homolog (yeast) ENSG00000075089 HU-07176-1 ACTR8 ARP8 actin-related protein 8 homolog (yeast) ENSG00000113812 HU-09411-1 AHCTF1 AT hook containing transcription factor 1 ENSG00000153207 HU-15150-1 AIRE autoimmune regulator ENSG00000160224 HU-12332-1 AKAP1 A kinase (PRKA) anchor protein 1 ENSG00000121057 HU-04065-1 ALG13 asparagine-linked glycosylation 13 homolog (S. cerevisiae) ENSG00000101901 HU-13552-1 ALKBH1 alkB, alkylation repair homolog 1 (E. coli) ENSG00000100601 HU-06662-1 ARID1A AT rich interactive domain 1A (SWI-like) ENSG00000117713 HU-12790-1 ARID1B AT rich interactive domain 1B (SWI1-like) ENSG00000049618 HU-09415-1 ARID2 AT rich interactive domain 2 (ARID, RFX-like) ENSG00000189079 HU-03890-1 ARID3A AT rich interactive domain 3A (BRIGHT-like) ENSG00000116017 HU-14677-1 ARID3B AT rich interactive domain 3B (BRIGHT-like) ENSG00000179361 HU-14203-1 ARID3C AT rich interactive domain 3C (BRIGHT-like) ENSG00000205143 HU-09104-1 ARID4A AT rich interactive domain 4A (RBP1-like) ENSG00000032219 HU-12512-1 ARID4B AT rich interactive domain 4B (RBP1-like) ENSG00000054267 HU-12520-1 ARID5A AT rich interactive domain 5A (MRF1-like) ENSG00000196843 HU-06595-1 ARID5B AT rich interactive domain 5B (MRF1-like) ENSG00000150347 HU-00556-1 ASF1A ASF1 anti-silencing function 1 homolog A (S.