Synthesis of Trialkyl 2-Halogeno-1,1,1-Ethanetricarboxylates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent (19) (11) 4,161,571 Yasui Et Al
United States Patent (19) (11) 4,161,571 Yasui et al. 45 Jul. 17, 1979 (54) PROCESS FOR PRODUCTION OF THE 4,080,493 3/1978 Yasui et al. .......................... 260/879 MALE CANHYDRDE ADDUCT OF A 4,082,817 4/1978 Imaizumi et al. ...................... 526/46 LIQUID POLYMER 4,091,198 5/1978 Smith ..................................... 526/56 75 Inventors: Seimei Yasui, Takarazuka; Takao FOREIGN PATENT DOCUMENTS Oshima, Sonehigashi, both of Japan 2262677 2/1975 France ....................................... 526/56 73) Assignee: Sumitomo Chemical Company, 44-1989 1/1969 Japan ......................................... 526/56 Limited, Osaka, Japan Primary Examiner-William F. Hamrock Attorney, Agent, or Firm-Birch, Stewart, Kolasch and 21 Appl. No.: 843,311 Birch 22 Filed: Oct. 18, 1977 57 ABSTRACT Related U.S. Application Data A process for production of the maleic anhydride ad duct of a liquid polymer having a maleic anhydride 62 Division of Ser. No. 733,914, Oct. 19, 1976, Pat, No. addition amount of 2 to 70% by weight, which com 4,080,493. prises reacting a liquid polymer having a molecular 51 Int. C.’................................................ CO8F 8/46 weight of 150 to 5,000 and a viscosity of 2 to 50,000 cp (52) U.S. C. ...................................... 526/90; 526/192; at 30 C. in the presence of at least one compound, as a 526/209; 526/213; 526/193; 526/195; 526/226; gelation inhibitor, selected from the group consisting of 526/233; 526/237; 526/238; 526/272; 525/285; imidazoles, thiazoles, metallic salts of mercapto 525/249; 525/251; 525/255; 525/245; 525/248 thiazoles, urea derivatives, naphthylamines, nitrosa (58) Field of Search ................ -

University Miaorilms International 300 N
INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1. The sign or “target” for pages apparently lacking from the document photographed is “Missing Page(s)”. If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obliterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed. For blurred pages, a good image of the page can be found in the adjacent frame. If copyrighted materials were deleted, a target note will appear listing the pages in the adjacent frame. 3. When a map, drawing or chart, etc., is part of the material being photographed, a definite method of “sectioning” the material has been followed. It is customary to begin filming at the upper left hand comer of a large sheet and to continue from left to right in equal sections with small overlaps. If necessary, sectioning is continued again—beginning below the first row and continuing on until complete. -

MI-GEE Brand Methylene Iodide
CHEMICAL BASED LIQUIDS BULLETIN NO.32 Chemical Based Products GEOLIQUIDS MI-GEE Brand Methylene Iodide MI-GEE is a pure form of Methylene Iodide and is widely moisture. Use in or exposure to strong sunlight or strong used for testing and separating minerals and for testing mercury vapor lights should be avoided. Any darkening is high index glasses, minerals and gems. It is also used in a due to liberation of a small amount of free iodine. This is variety of organic chemical syntheses. One of these is the easily removed by shaking with a cool 5 - 10% solution of Simmons & Smith Synthesis (JACS 80, 5323, 1958) for Sodium Hydroxide or Sodium Carbonate, washing twice with putting a methylene group into an organic molecule. plain water, separating and filtering. Due to the highly labile nature of Methylene Iodide, Gas Chromatography can not be Properties: used to analyze it, as heat decomposes the material to give low results. MI-GEE is shipped with Copper wire screen pre- MI-GEE Assays 99.9% Minimum CH2I2 servative added. Copper wire sinks to the bottom, causes Formula Weight: 267:87 no difficulty in use, does not contaminate MI-GEE, and will keep it light in color for several years. MI-GEE should always Synonym: Diiodomethane be stored in total darkness and in closed containers when Iodine Content, Theory: 94.76% not in use. Mercury should never be used to lighten the color or preserve Methylene Iodide as it produces toxic Density: d2 0= 3.325 (Very close to Specific Gravity) 4 compounds. 15 Refractive Index: n D = 1.74 Melting Point: 5-6°C (4l - 42.8°F) MI-GEE Is miscible with acetone, methanol, ethanol, methy- lene chloride, ether, chloroform, dimethyl sulfoxide and Boiling Point: 181°C (358ºF) other solvents. -

Preparation and 13C NMR Investigation of 1,1-Dilithioalkenes
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1990 Preparation and 13C NMR investigation of 1,1-dilithioalkenes, 1,2-dilithioalkenes, 1-lithiocyclopropenes and 1,2-dilithiocyclopropenes Hon-Wah Man Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Man, Hon-Wah, "Preparation and 13C NMR investigation of 1,1-dilithioalkenes, 1,2-dilithioalkenes, 1-lithiocyclopropenes and 1,2-dilithiocyclopropenes " (1990). Retrospective Theses and Dissertations. 9860. https://lib.dr.iastate.edu/rtd/9860 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. MICROFILMED 1991 INFORMATION TO USERS The most advanced technology has been used to photograph and reproduce this manuscript from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

Intermolecular Interaction in Methylene Halide (CH2F2, Ch2cl2, Ch2br2 and CH2I2) Dimers
molecules Article Intermolecular Interaction in Methylene Halide (CH2F2, CH2Cl2, CH2Br2 and CH2I2) Dimers László Almásy 1,2,* ID and Attila Bende 3,* ID 1 State Key Laboratory of Environment-friendly Energy Materials, Southwest University of Science and Technology, Mianyang 621010, China 2 Institute for Solid State Physics and Optics, Wigner Research Centre for Physics, Konkoly Thege út 29-33, 1121 Budapest, Hungary 3 Molecular and Biomolecular Physics Department, National Institute for Research and Development of Isotopic and Molecular Technologies, Donat Street, No. 67-103, Ro-400293 Cluj-Napoca, Romania * Correspondence: [email protected] (L.A.); [email protected] (A.B.) Academic Editors: Igor Reva and Xuefeng Wang Received: 27 April 2019; Accepted: 1 May 2019; Published: 10 May 2019 Abstract: The intermolecular interaction in difluoromethane, dichloromethane, dibromomethane, and diiodomethane dimers has been investigated using high level quantum chemical methods. The potential energy curve of intermolecular interaction along the C··· C bond distance obtained using the coupled-cluster theory with singles, doubles, and perturbative triples excitations CCSD(T) were compared with values given by the same method, but applying the local (LCCSD(T)) and the explicitly correlated (CCSD(T)-F12) approximations. The accuracy of other theoretical methods—Hartree–Fock (HF), second order Møller–Plesset perturbation (MP2), and dispersion corrected DFT theory—were also presented. In the case of MP2 level, the canonical and the local-correlation cases combined with the density-fitting technique (DF-LMP2)theories were considered, while for the dispersion-corrected DFT, the empirically-corrected BLYP-D and the M06-2Xexchange-correlation functionals were applied. In all cases, the aug-cc-pVTZ basis set was used, and the results were corrected for the basis set superposition error (BSSE) using the counterpoise method. -

United States Patent Office Patented Dec
2,964,508 United States Patent Office Patented Dec. 13, 1960 2 diiodomethane, iodotribromomethane, chlorotriiodometh 2,964,508 ane, dichlorodiiodomethane, iodotrichloromethane, fluo rotriiodomethane, difluorodiiodomethane, and iodotri PROCESS FOR CONTROLLING THE MOLECU. fluoromethane. LAR WEIGHT OF VENYLDENE CHLORIDE 5 The amount of the perhalomethane that may be em POLYMERS ployed in the process of this invention may be varied Marion R. Rector and William E. Cohrs, Midland, Mich., from 0.05 to 5.0 percent by weight based on the weight assignors to The Dow Chemical Company, Midland, of the monomer. The preferred concentration is from Mich., a corporation of Delaware 1 to 3 percent by weight based on the weight of the No Drawing. 0 monomer. When less than 0.05 percent is used the Filed June 23, 1955, Ser. No. 517,663 amount of molecular weight lowering is not usually great 6 Claims. (C. 260-87.7) enough for practical purposes. When more than 5.0 percent is used no additional benefits are realized. When the compounds are used in these concentrations a sub This invention relates to a process for controlling the 5 stantial lowering of molecular weight of the polymers molecular weight of polymers. More particularly, it re occurs without the necessity of altering polymerization lates to a process for preparing vinylidene chloride poly conditions, and without significant decrease in polymeri mers and copolymers having lower molecular weights zation rate. than similar polymers prepared at the same tempera Any of the known methods of polymerization may be tures but using prior known processes. 20 employed in this process (mass, aqueous emulsion, aque Vinylidene chloride polymers and copolymers of viny ous suspension, or solution), although especially ad lidene chloride with another copolymerizable monomer vantageous results are obtained when the common non such as vinyl chloride have been fabricated into a wide emulsified aqueous suspension method is used. -
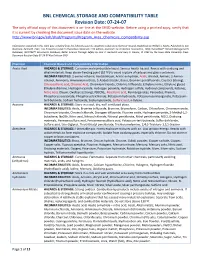
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Download (6MB)
STUDIES ON THE GENERATION AND REACTIVITY OF METAL CARBENOIDS A Thesis Presented by Rehan Aqil In Partial Fulfilment of the Requirements for the Award of the Degree of DOCTOR OF PHILOSOPHY OF THE UNIVERSITY OF LONDON Christopher Ingold Laboratories Department of Chemistry University of London London WCIH OAJ September 2002 ProQuest Number: U643094 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest U643094 Published by ProQuest LLC(2015). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract ABSTRACT This thesis concerns the generation of metal carbenoids, in particular organozinc carbenoids, and their subsequent reactions, such as C-H insertion reactions or cyclopropanation with alkenes. The thesis is divided into three major sections. The introductory chapter presents a comparative review covering metal carbenoid species. The review focuses on how their reactivity is controlled by the metal, the leaving group and by the nature of electron-donating and -withdrawing groups on the carbenoid and finally which type of alkenes give the highest reactivity and yields as a function of these parameters. The results and discussion chapter opens with a brief overview of the organozinc carbenoid chemistry which has been developed within our group. -

Iodoform - Wikipedia
6/13/2020 Iodoform - Wikipedia Iodoform Iodoform (also known as triiodomethane and, inaccurately, as carbon triiodide) is the organoiodine compound with the Iodoform formula CHI3. A pale yellow, crystalline, volatile substance, it has a penetrating and distinctive odor (in older chemistry texts, the smell is sometimes referred to as that of hospitals, where the compound is still commonly used) and, analogous to chloroform, sweetish taste. It is occasionally used as a disinfectant. Contents Structure Synthesis and reactions Natural occurrence Applications Names See also Preferred IUPAC name Triiodomethane References Other names External links Iodoform;[1] Carbon triiodide Structure Identifiers CAS Number 75-47-8 (http://w The molecule adopts tetrahedral molecular geometry with C3v ww.commonche symmetry. mistry.org/Chemi calDetail.aspx?re Synthesis and reactions f=75-47-8) 3D model (JSmol) Interactive image The synthesis of iodoform was first described by Georges-Simon (https://chemapp Serullas in 1822, by reactions of iodine vapour with steam over red- s.stolaf.edu/jmol/j hot coals, and also by reaction of potassium with ethanolic iodine in mol.php?model=I the presence of water;[5] and at much the same time independently C%28I%29I) by John Thomas Cooper.[6] It is synthesized in the haloform reaction by the reaction of iodine and sodium hydroxide with any Beilstein 1697010 one of these four kinds of organic compounds: a methyl ketone Reference (CH3COR), acetaldehyde (CH3CHO), ethanol (CH3CH2OH), and ChEBI CHEBI:37758 (ht certain secondary alcohols (CH3CHROH, where R is an alkyl or aryl tps://www.ebi.ac. group). uk/chebi/searchI d.do?chebiId=37 758) ChEMBL ChEMBL1451116 (https://www.ebi. -
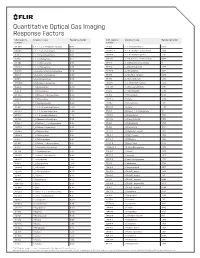
Quantitative Optical Gas Imaging Response Factors CAS Registry Chemical Name Response Factor† CAS Registry Chemical Name Response Factor† Number* Number*
Quantitative Optical Gas Imaging Response Factors CAS registry Chemical name Response factor† CAS registry Chemical name Response factor† number* number* 431-89-0 1_1_1_2_3_3_3-Heptafluoropropane 0.103 75-89-8 2_2_2-Trifluoroethanol 0.326 630-20-6 1_1_1_2-Tetrachloroethane 0.132 25256-77-4 2_2_4-Trimethyl-1_3-pentanediol 1.086 75-68-3 1_1_1-Chlorodifluoroethane 0.117 107-40-4 2_2_4-Trimethyl-2-pentene 1.262 71-55-6 1_1_1-Trichloroethane 0.121 306-83-2 2_2-Dichloro-1_1_1-trifluoroethane 0.047 421-50-1 1_1_1-Trifluoroacetone 0.086 76-11-9 2_2-Difluorotetrachloroethane 0 420-46-2 1_1_1-Trifluoroethane 0.106 75-83-2 2_2-Dimethyl_butane 1.944 354-14-3 1_1_2_2-Tetrachloro-1-fluoroethane 0.046 431-03-8 2_3-Butanedione 0.274 79-34-5 1_1_2_2-Tetrachloroethane 0.069 78-88-6 2_3-Dichloro-1-propene 0.279 79-00-5 1_1_2-Trichloroethane 0.102 79-29-8 2_3-Dimethylbutane 1.014 1717-00-6 1_1-Dichloro-1-fluoroethane 0.12 107-39-1 2_4_4-Trimethyl-1-pentene 1.212 75-34-3 1_1-Dichloroethane 0.272 584-84-9 2_4-Diisocyanatetoluene 0.895 75-35-4 1_1-Dichloroethene 0.017 87-62-7 2_6-Dimethylaniline 1.308 471-43-2 1_1-Difluoro-2_2-dichloroethane 0.352 75-26-3 2-Bromopropane 0.733 73-37-6 1_1-Difluoroethane 0.535 107-01-7 2-Butene 1.204 57-14-7 1_1-Dimethylhydrazine 0.897 111-76-2 2-Butoxyethanol 1.962 119-64-2 1_2_3_4-Tetrahydronaphthalene 2.439 554-61-0 2-Carene 1.286 488-23-3 1_2_3_4-Tetramethylbenzene 1.484 75-88-7 2-Chloro-1_1_1-trifluoroethane 0.1 527-53-7 1_2_3_5-Tetramethylbenzene 1.402 107-07-3 2-Chloroethanol 0.593 124-73-2 1_2-Dibomotetrafluoroethane -

Solvation Effects on the A-Band Photodissociation of Dibromomethane: Turning a Photodissociation Into a Photoisomerization†
10464 J. Phys. Chem. A 2000, 104, 10464-10470 ______________________________________________________________________________________http:\\www.paper.edu.cn Solvation Effects on the A-Band Photodissociation of Dibromomethane: Turning a Photodissociation into a Photoisomerization† Xuming Zheng, Wai Ming Kwok,‡ and David Lee Phillips* Department of Chemistry, The UniVersity of Hong Kong, Pokfulam Road, Hong Kong ReceiVed: March 28, 2000; In Final Form: July 18, 2000 We have obtained A-band resonance Raman spectra of dibromomethane in the gas and solution phase and nanosecond time-resolved resonance Raman spectra of dibromomethane photoproducts. The A-band resonance Raman spectra suggest the short-time dynamics are similar to other dihalomethanes that are known to have direct photodissociation reactions in the gas phase. Several power dependent A-band resonance Raman bands were tentatively assigned to the C-Br stretch overtone progression of the CH2Br radical which has a strong absorption band that is coincident with the A-band resonance Raman excitation wavelength. Two-color nanosecond time-resolved resonance Raman spectra (266.0 nm pump/341.5 nm probe) were obtained and comparison of the vibrational frequencies to the results for density functional theory calculations indicate that the iso-CH2Br-Br species is mainly responsible for the transient photoproduct absorption band ∼360 nm. Our preliminary results for A-band photoexcitation of dibromomethane in conjunction with previously reported diiodomethane results suggest that solvation effects -

潤滑添加劑로서의 A^-Iodopyridinium Dichlorodate 의 화학반응성 文 卓 珍•權 五 昇 高麗大學校理工大學 材料工學科 (1974
DAEHAN HWAHAK HWOEJEE (Journal of the Korean Chemical Society) Vol. 19, No.l, 1975 Printed in Republic of Korea 潤滑添加劑로서의 A^-Iodopyridinium Dichlorodate 의 화학반응성 文 卓 珍•權 五 昇 高麗大學校理工大學 材料工學科 (1974. 8. 29 접수 ) Chemical Reactivity of 7V-Iodopyridinium Dichlorodate as a Lubricant Additive Tak Jin Moon and Oh Seung Kwon Department of Materials Science Korea Universityi Seoul, Korea (Received Aug. 29, 1974) 요 약 소량의 요오드 화합물이 윤활유에 함유될 때 금속표면사이의 마찰을 감소시 키 고 내 마모성 을 향상시키 는데 이는 금속표면에 박층상구조의 이요오드화물이 형성되기 때문이 라고 한다 . 그러 나 금속표면에서의 화학반응성 , 마모와 극압실험 및 hot wire method 에 의한 결과에 의하면 윤활기구 는 이 요오드화물의 박층상 구조가 아니고 다른 기구에 의한 것임을 알았다 . 유기 요오드화합 물중에 서 특히 A^-iodopyridinium dichlorodate 는 double charge transfer complex 로써 다른 methyl iodide 계 의 화합물 보다 금속표면 사이 의 마찰을 더 욱 감소시 키 고 윤활기 구는 interhalogen 의 화학반응에 의한 것임을 알았다 . Abstract Small amounts of iodine compound in mineral oils are usually effective in reducing friction of metallic surfaces. Such improvement in frictional behaviour of wear 산 】aracteristics was explained by the formation of a diiodide layer lattice structure at the metallic contact surfaces. The lubrication mechanism, however, by which organoiodine compounds functions is not based on the formation of such lattice structure iodide. It was tested and shown, by a static surface chemical reactivity test, wear and EP tests, and a hot wire methcxl, that compound such as Ar-iodopyridinium dichlorodate, a double charge transfer complex, reacted with metals as an interhalogen compound and that the resultant thin film product reduced appreciable the friction of metallic surfaces, more than compounds such as methyl iodide, diiodomethane, and iodoform.