Differences in Gut Microbial Diversity Are Driven by Drug Use and Drug
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Comparative Study of Livestock's Name Between Ancient And
US-China Foreign Language, ISSN 1539-8080 September 2014, Vol. 12, No. 9, 728-735 D DAVID PUBLISHING A Comparative Study of Livestock’s Name Between Ancient and Modern Yi Language of China∗ YANG Liu-jin Honghe University, Mengzi City, China The traditional pronunciations of domestic animal lexemes recorded in ancient Nisu(Yi) manuscripts differ significantly from the vernacular pronunciations of their modern counterparts. Thus, in the process of translating such texts, when a translator substitutes modern Nisu for ancient Nisu, not only can many of the characteristics of the original text be lost, but the original meaning may also be completely modified. Currently a number of specialists and scholars, both in China and elsewhere, are familiar with modern Nisu but have no such familiarity with the language’s ancient forms. The paper grants Nisu researchers a better grasp of the differences between the language’s ancient and modern forms in order to help others avoid mistakes in translating ancient Nisu manuscripts. This is accomplished through a brief comparative analysis of Nisu names for livestock and fowl—then and now. Keywords: Yi Language, livestock, name Introduction Nisu is a Tibet-Burman language of the Ngwi branch which has been officially classified as an ethnic linguistic sub-branch of the Yi nationality in China. The Nisu retain use of their language in both spoken and written forms. The current population of the official Yi nationality stands at 7,776,230 (PCPC, 2002) and is distributed through Yunnan, Sichuan, Guangxi, and Guizhou Provinces. Traditionally, in China, the languages spoken by the Yi nationality have been divided into six major dialect regions: Northern, Eastern, Southern, Western, Central, and Southeastern (YYJS, 1987, pp. -

County, Province 包装厂中文名chinese Name of Packing House
序号 注册登记号 所在地 Location: 包装厂中文名 包装厂英文名 包装厂中文地址 包装厂英文地址 Numbe Registered Location County, Province Chinese Name of Packing house English Name of Packing house Address in Chinese Address in English r Number 1 北京平谷 PINGGU,BEIJING 北京凤凰山投资管理中心 BEIJING FENGHUANGSHAN INVESTMENT MANAGEMENT CENTER 平谷区峪口镇 YUKOU,PINGU DISTRICT,BEIJING 1100GC001 2 北京平谷 PINGGU,BEIJING 北京东四道岭果品产销专业合作社 BEIJING DONGSIDAOLING FRUIT PRODUCTION AND MARKETING PROFESSIONNAL COOPERATIVES平谷区镇罗营镇 ZHENLUOYING,PINGGU DISTRICT,BEIJING 1100GC002 TIANJIN JIZHOU DEVELOPMENT ZONE, WEST IN ZHONGCHANG SOUTH ROAD, NORTH 3 天津蓟州区 JIZHOU,TIANJIN 天津蓟州绿色食品集团有限公司 TIANJIN JIZHOU GREEN FOOD GROUP CO., LTD. 天津市蓟州区开发区中昌南路西、京哈公路北IN JING-HA ROAD 1200GC001 4 河北辛集 XINJI,HEBEI 辛集市裕隆保鲜食品有限责任公司果品包装厂XINJI YULONG FRESHFOOD CO.,LTD. PACKING HOUSE 河北省辛集市南区朝阳路19号 N0.19 CHAOYANG ROAD, SOUTH DISTRICT OF XINJI CITY, HEBEI PROVINCE 1300GC001 5 河北辛集 XINJI,HEBEI 河北天华实业有限公司 HEBEI TIANHUA ENTERPRISE CO.,LTD. 河北省辛集市新垒头村 XINLEITOU VILLAGE,XINJI CITY,HEBEI 1300GC002 6 河北晋州 JINZHOU,HEBEI 河北鲜鲜农产有限公司 HEBEI CICI CO., LTD. 河北省晋州市工业路33号 NO.33 GONGYE ROAD,JINZHOU,HEBEI,CHINA 1300GC004 7 河北晋州 JINZHOU,HEBEI 晋州天洋贸易有限公司 JINZHOU TIANYANG TRADE CO,. LTD. 河北省晋州市通达路 TONGDA ROAD, JINZHOU CITY,HEBEI PROVINCE 1300GC005 8 河北晋州 JINZHOU,HEBEI 河北省晋州市长城经贸有限公司 HEBEI JINZHOU GREAT WALL ECONOMY TRADE CO.,LTD. 河北省晋州市马于开发区 MAYU,JINZHOU,HEBEI,CHINA 1300GC006 9 河北晋州 JINZHOU,HEBEI 石家庄市丰达金润农产品有限公司 SHIJIAZHUANG GOLDEN GLORY AGRICULTURAL CO.,LTD. 晋州市马于镇北辛庄村 BEIXINZHUANG,JINZHOU,HEBEI,CHINA 1300GC007 10 河北赵县 ZHAO COUNTY,HEBEI 河北嘉华农产品有限责任公司 HEBEI JIAHUA -
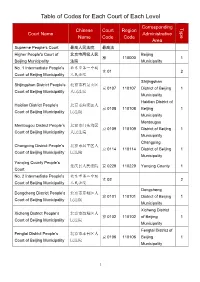
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Save the Children in China 2013 Annual Review
Save the Children in China 2013 Annual Review Save the Children in China 2013 Annual Review i CONTENTS 405,579 In 2013, Save the Children’s child education 02 2013 for Save the Children in China work helped 405,579 children and 206,770 adults in China. 04 With Children and For Children 06 Saving Children’s Lives 08 Education and Development 14 Child Protection 16 Disaster Risk Reduction and Humanitarian Relief 18 Our Voice for Children 1 20 Media and Public Engagement 22 Our Supporters Save the Children organised health and hygiene awareness raising activities in the Nagchu Prefecture of Tibet on October 15th, 2013 – otherwise known as International Handwashing Day. In addition to teaching community members and elementary school students how to wash their hands properly, we distributed 4,400 hygiene products, including washbasins, soap, toothbrushes, toothpastes, nail clippers and towels. 92,150 24 Finances In 2013, we responded to three natural disasters in China, our disaster risk reduction work and emergency response helped 92,150 Save the Children is the world’s leading independent children and 158,306 adults. organisation for children Our vision A world in which every child attains the right to survival, protection, development and 48,843 participation In 2013, our child protection work in China helped 48,843 children and 75,853 adults. Our mission To inspire breakthroughs in the way the world treats children, and to achieve immediate and 2 lasting change in their lives Our values 1 Volunteers cheer on Save the Children’s team at the Beijing Marathon on October 20th 2013. -

Original Article Galectin-1 and DNMT1 Are Highly Expressed and Related to the Prognoses of Patients with Cervical Cancer
Int J Clin Exp Med 2020;13(3):1439-1446 www.ijcem.com /ISSN:1940-5901/IJCEM0103882 Original Article Galectin-1 and DNMT1 are highly expressed and related to the prognoses of patients with cervical cancer Xiaomin Guo1, Chunyan Yang2, Hongying Zhao3, Xingjun Wu4, Yanling Dai5 1School of Nursing, Chuxiong Medical and Pharmaceutical College, Chuxiong City, Yunnan Province, China; 2Department of Pharmacy, Chuxiong Medical and Pharmaceutical College, Chuxiong City, Yunnan Province, China; 3Department of Tumor and Hematology, The First People’s Hospital, Honghe Prefecture, Mengzi City, Yunnan Province, China; 4Qujing Medical College, Qujing City, Yunnan Province, China; 5Department of Laboratory, Chuxiong Medical and Pharmaceutical College, Chuxiong City, Yunnan Province, China Received October 23, 2019; Accepted November 21, 2019; Epub March 15, 2020; Published March 30, 2020 Abstract: Objective: To explore the expressions of galectin-1 and human DNA methyltransferase 1 (DNMT1) in cervical cancer tissues and normal cervical tissues and their clinical significance. Method: Eighty-four patients with cervical cancer were randomly selected. 84 samples of cancer tissues and their para-carcinoma tissues were ana- lyzed with regard to the following parameters: Galectin-1 and DNMT1 mRNA analyzed by RT-PCR, and galectin-1 and DNMT1 protein by western blotting. The relationships between the expressions of galectin-1 and DNMT1 protein, the clinicopathological factors, and the prognoses were analyzed. A correlation analysis was performed between the galectin-1 and DNMT1 protein expressions in the cervical cancer tissues. Result: The expressions of galectin-1 and DNMT1 mRNA and protein in the cervical cancer tissues were higher than they were in the para-carcinoma tis- sues (P<0.05). -

Prevalence of Four Common Bee RNA Viruses in Eastern Bee
ary Scien in ce r te & e T V e f c h o Journal of Veterinary Science & n n l o o a a l l n n o o r r g g u u Wang, et al., J Veterinar Sci Technol 2016, 7:1 y y o o J J Technology DOI: 10.4172/2157-7579.1000284 ISSN: 2157-7579 Research Article Open Access Prevalence of Four Common Bee RNA Viruses in Eastern Bee Populations in Yunnan Province, China Miao Wang1, Junlong Bi2,3, Lei Wang1,3, Danyin Zhou1, Xiaotian Ma1, Wengui Li2, Wenzheng Zhao1, Gefen Yin2, Jianping Liu2,4*, Shaoyu He1* 1Eastern Bee Research Institute, Yunnan Agricultural University, Kunming 650201, China 2College of Animal Science and Technology, Yunnan Agricultural University, Kunming 650201, China 3Chuxiong Prefecture Animal Disease Control Center, Chuxiong 675000, Yunnan Province, China 4Karolinska Institute, Department of Biosciences and Nutrition, SE-14183 Huddinge, Sweden *Corresponding author: Jianping Liu, Karolinska Institute, Department of Biosciences and Nutrition, SE-14183 Huddinge, Sweden, Tel: 858586658; E-mail: [email protected] Shaoyu He, Eastern Bee Research Institute, Yunnan Agricultural University, Kunming 650201, China, Email: [email protected] Rec date: Dec 17, 2015; Acc date: Jan 11, 2016; Pub date: Jan 13, 2016 Copyright: © 2016 Wang M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Deformed wing virus (DWV), black queen cell virus (BQCV), sacbrood virus (SBV), Kashmir bee virus (KBV), acute bee paralysis virus (ABPV) and chronic bee paralysis virus (CBPV) are the most common RNA viruses in bee population worldwide. -

China's Cities
THE STATE OF THE STATE THE STATE OF CHINA’S CITIES CHINA’S CITIES CHINA’S 2014/2015 2014/2015 THE STATE OF CHINA’S CITIES 2014/2015 SPONSOR International Eurasian Academy of Sciences UNDERTAKER China Science Center of International Eurasian Academy of Sciences CO-ORGANIZERS China Association of Mayors Urban Planning Society of China EDIROR-IN-CHIEF Wang Guangtao, Secretary-General, International Eurasian Academy of Sciences (IEAS), Executive Vice President, China Science Center of International Eurasian Academy of Sciences (CSC-IEAS) HONORARY EDITOR-IN-CHIEF Tao Siliang, Executive Vice President, China Association of Mayors EXECUTIVE EDITOR-IN-CHIEF Mao Qizhi, Academician, IEAS, Professor, School of Architecture, Tsinghua University Shao Yisheng, Academician, IEAS, Deputy Secretary-General, CSC-IEAS AUTHORS Mao Qizhi, Academician, IEAS, Professor, School of Architecture, Tsinghua University Shao Yisheng, Academician, IEAS, Researcher, Vice President, China Academy of Urban Planning and Design Shi Nan, Professor, Secretary-General, Urban Planning Society of China Shen Jianguo, PhD., Inter-Regional Adviser, United Nations Human Settlements Programme Yu Taofang, PhD., Associate Professor, School of Architecture, Tsinghua University Zhang Zhiguo, PhD., Associate Researcher, China Academy of Urban Planning and Design Li Lin, Associate Senior Editor, City Planning Review magazine Chen Xiaohui, Deputy Chief Planner, Jiangsu Institute of Urban Planning and Design Qu Changhong, Senior Engineer, Deputy Secretary-General, Urban Planning Society of -

参展商名录 Exhibitor List
参展商名录 EXHIBITOR LIST ◆ 1 号馆·旅游馆 Hall #1: Tourism Pavilion…………………………………………………………… 1 ◆ 2 号馆·农业馆 Hall #2: Agriculture Pavilion ……………………………………………………… 6 ◆ 3 号馆·制造业馆 Hall #3: Manufacturing Industry Pavilion ………………………………………… 26 ◆ 4 号馆·新材料馆 Hall #4: New Materials Pavilion …………………………………………………… 35 ◆ 5 / 6 号馆·东南亚馆 Hall #5 & 6: Southeast Asia Pavilions ……………………………………………… 38 ◆ 8 / 9 号馆·南亚馆 Hall #8 & 9: South Asia Pavilions ………………………………………………… 64 ◆ 10 / 11 号馆·境外馆 Hall #10 & 11: Overseas Pavilions ………………………………………………… 84 ◆ 12 号馆·台湾馆 Hall #12 : Taiwan Pavilion ……………………………………………………… 105 ◆ 13 号馆·境内馆 Hall #13 : Domestic Pavilion …………………………………………………… 116 ◆ 14 号馆·食尚生活馆 Hall #14 : Food and Delicacies Pavilion ………………………………………… 138 ◆ 15 号馆·机电馆 Hall #15 : Mechatronics Pavilion ………………………………………………… 142 ◆ 16 号馆·生物医药与大健康馆 Hall #16 : Biomedicine and Health Pavilion …………………………………… 149 ◆ 17 号馆·文化创意馆 Hall #17 : Culture Creativity Pavilion …………………………………………… 155 ◆ 18 / 19 号馆·木艺馆 Hall #18 & 19 : Wood Culture Pavilions ………………………………………… 160 第三层展馆分布 3F rd Layout of the 3 floor 7号馆 8号馆 6号馆 开及 幕主 主 大 宾 题 厅 国 国 馆 东 南 亚 馆 南 亚 馆 9号馆 5号馆东 南 亚 馆 南 亚 馆 10号馆 4号馆新 材 料 馆 境 外 馆 11号馆 制 造 业 馆 境 外 馆 3号馆 12号馆 农业馆 台 湾 馆 2号馆 13号馆 境 内 馆 旅游馆 1号馆 第三层共设13个展馆,展览面积13万平方米。 The 3 rd floor of the exhibition center will have 13 pavilions with exhibition area of 130,000 . Pavilion 1 旅游馆 Tourism Pavilion Pavilion 2 农业馆 Agriculture Pavilion Pavilion 3 制造业馆 Manufacturing Industry Pavilion Pavilion 4 新材料馆 New Materials Pavilion Pavilion 5/6 东南亚馆 Southeast Asia Pavilions Pavilion 7 开幕大厅及主题国主宾国馆 Ceremonial & -

Download.Atlantis-Press.Com
Advances in Social Science, Education and Humanities Research (ASSEHR), volume 80 International Conference on Education, Culture and Social Development (ICECSD 2017) A Brief Analysis of the Cultural Connotation and Characteristics of the Urban Sculpture Culture of Mengzi City Zong.Yan Honghe.University.Yunnan.China Email:[email protected] Abstract: Mengzi is a developing medium-sized city with long history and massive culture. Along with the development, the image of the city has got ascended rapidly. The city’s sculpture has got certain development. Mengzi is a culture famous city in southwest China with many the firsts of Yunnan Province. The outstanding characteristic cultures are: the French concession culture, the Southwest Associated University culture, the Yunnan Rice Noodles culture and the pomegranate culture. Most urban sculptures are around those important cultures. By looking up to the documents, and making fact-finding survey , the state-of-the-art, the distribution and the variety of the sculptures in Mengzi are analyzed. Some typical cases are analyzed and compared. The cultural features are considered to analyze the art characteristic and cultural connotation of the sculptures in Mengzi. Key words: Mengzi City; urban sculpture; cultural connotation; art feature 1 A Survey of the Urban Sculpture in Mengzi 1.1The Culture Background of Mengzi Mengzi is an important town in southern Yunnan, which has a long history. It has abundant resources and prosperous culture. In the history of more than 2000 years, it sets multiculture, such as the borderland culture, the central plain culture, the Party’s history culture, the brigade culture, and the western culture as a rich ore with thick historical accumulation and various cultural resources. -

Cash Crops and Climate Shocks: Flexible Livelihoods in Southeast Yunnan, China
CASH CROPS AND CLIMATE SHOCKS: FLEXIBLE LIVELIHOODS IN SOUTHEAST YUNNAN, CHINA Clara CHAMPALLE Department of Geography McGill University, Montreal Submitted December 2012 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master of Arts © Clara Champalle 2012 ABSTRACT The rural landscape of the People’s Republic of China has changed dramatically from land collectivization in the 1950s to the decollectivization reforms initiated by Deng Xiaoping in 1979. By the mid-1980s each rural household had again become responsible for its own agricultural production, and food security began to improve, even within the most remote areas. To further this agrarian transition, in the late 1990s the central state devised the Western Development Strategy to advance its ‘less developed’ western regions, within which provincial governments subsidized cash crops. The aim of this thesis is first to examine the importance of cash crops and related subsidies for Han and minority nationality farmer households in Honghe Hani-Yi Autonomous Prefecture, Yunnan, China; second, to assess how extreme weather events affect these farmers’ livelihoods and to investigate the coping mechanisms they employ. To answer this aim I draw on a conceptual framework that incorporates key elements from sustainable livelihoods, food security, and vulnerability and resilience to climate variability literatures. Focusing on four townships in Honghe Prefecture, southeast Yunnan, I completed statistical analyses of quantitative data regarding recent extreme weather events in the region and ethnographic fieldwork, including conversational interviews with farmers and semi-structured interviews with local officials completed in summer 2011. I find that state-sponsored cash crops do not always bring higher financial capital rewards and that cash crop farmers have been increasingly exposed to extreme precipitation and temperatures since the year 2000, which constrain their access to livelihood capitals, essential for (re)investing in cash cropping. -

Project Information Document
PROJECT INFORMATION DOCUMENT (PID) APPRAISAL STAGE Report No.: PIDA2551 Public Disclosure Authorized Project Name CN Yunnan Honghe Prefecture Diannan Center Urban Transport (P101525) Region EAST ASIA AND PACIFIC Public Disclosure Copy Country China Sector(s) Urban Transport (90%), General transportation sector (10%) Theme(s) City-wide Infrastructure and Service Delivery (100%) Lending Instrument Investment Project Financing Project ID P101525 Borrower(s) People's Republic of China Implementing Agency Honghe Prefecture Government Public Disclosure Authorized Environmental Category B-Partial Assessment Date PID Prepared/Updated 17-Jan-2014 Date PID Approved/Disclosed 21-Jan-2014 Estimated Date of Appraisal 30-Dec-2013 Completion Estimated Date of Board 15-May-2014 Approval Decision I. Project Context Public Disclosure Authorized Country Context During the past three decades, China has experienced unprecedented economic growth, accompanied by rapid urbanization. According to the National Statistics Bureau, China’s urban population reached 712 million by the end of 2012, accounting for 52.6 percent of the total Public Disclosure Copy population. The United Nations projects that the number of Chinese urban dwellers will grow to nearly 1 billion by 2030, accounting for 20 percent of the total increase in urban population worldwide. Economic growth and urbanization, however, have taken place disproportionally in different regions of China – for instance, per capita Gross Domestic Product (GDP) in the eastern provinces and the western provinces were RMB 53,350 (US$ 8,746 equivalent) and RMB 27,731 (US$ 4,546 equivalent), respectively, and the percentages of urban population were 60.8 percent and 43.0 percent, respectively, in 2011. Efforts to narrow the development gaps between the western region Public Disclosure Authorized and the eastern region were initiated in 2000 when the central government of China launched the Western Regional Development (WRD) Program. -

Urban Development in the Greater Mekong Subregion
URBAN DEVELOPMENT IN THE GREATER MEKONG SUBREGION ASIAN DEVELOPMENT BANK URBAN DEVELOPMENT IN THE GREATER MEKONG SUBREGION Edited by Florian Steinberg and Januar Hakim ASIAN DEVELOPMENT BANK Creative Commons Attribution 3.0 IGO license (CC BY 3.0 IGO) © 2016 Asian Development Bank 6 ADB Avenue, Mandaluyong City, 1550 Metro Manila, Philippines Tel +63 2 632 4444; Fax +63 2 636 2444 www.adb.org; openaccess.adb.org Some rights reserved. Published in 2016. Printed in the Philippines. ISBN 978-92-9254-983-1 (Print), 978-92-9254-984-8 (e-ISBN) Publication Stock No. BKK146799 Cataloging-In-Publication Data Asian Development Bank. Urban development in the Greater Mekong Subregion. Mandaluyong City, Philippines: Asian Development Bank, 2016. 1. Urban development. 2. Regional development. 3. Greater Mekong Subregion. I. Asian Development Bank. The views expressed in this publication are those of the authors and do not necessarily reflect the views and policies of the Asian Development Bank (ADB) or its Board of Governors or the governments they represent. ADB does not guarantee the accuracy of the data included in this publication and accepts no responsibility for any consequence of their use. The mention of specific companies or products of manufacturers does not imply that they are endorsed or recommended by ADB in preference to others of a similar nature that are not mentioned. By making any designation of or reference to a particular territory or geographic area, or by using the term “country” in this document, ADB does not intend to make any judgments as to the legal or other status of any territory or area.