Statistical Essays Motivated by Genome-Wide Association Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Yan Et Al. Supplementary Material
SUPPLEMENTARY MATERIAL FOR: CELL ATLAS OF THE HUMAN FOVEA AND PERIPHERAL RETINA Wenjun Yan*, Yi-Rong Peng*, Tavé van Zyl*, Aviv Regev, Karthik Shekhar, Dejan Juric, and Joshua R, Sanes^ *Co-First authors ^Author for correspondence, [email protected] Figure S1 tSNE visualization showing contributions to cell types by batch for photoreceptors (a), horizontal cells (b), bipolar cells (c), amacrine cells (d), retinal ganglion cells (e) and non-neuronal cells (f). Each dot represents one cell. Colors distinguish retina samples. Source of each sample is shown in Table S1. Overall, batch eFFects were minimal. Figure S2 Violin and superimposed box plots showing expression of OPN4 in RGC clusters Figure S3 Heat maps showing expression patterns of disease genes by cell classes in the Fovea and periphery. Only genes expressed by more than 20% of cells in any individual class in either Fovea or peripheral cells are plotted. Table S1 Information on donors from whom retinal cells were obtained for scRNA-seq proFiling. Table S2 Publications reporting single cell or single nucleus profiling on cells from human retina. 1 Figure S1 a b c PR HC BP H1 H2F1 H2F2 H3 tSNE1 tSNE1 H4 tSNE1 H5 H9 H11 tSNE2 tSNE2 tSNE2 e AC f RGC g Non-neuronal tSNE1 tSNE1 tSNE1 tSNE2 tSNE2 tSNE2 Figure S2 OPN4 4 2 log Expression 0 MG-ON MG-OFF PG-OFF PG-ON hRGC5 hRGC6 hRGC7 hRGC8 hRGC9 hRGC10 hRGC11 hRGC12 Figure S3 Fovea Fovea Peripheral Peripheral Rods Cones BP HC AC RGC Muller Astro MicG Endo Rods Cones BP HC AC RGC Muller Astro MicG Endo ARL13B MAPK8IP3 EXOC6 LSM4 Expression -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Prenatal Testing Requisition Form
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 PRENATAL COMPREHENSIVE REQUISITION FORM PATIENT INFORMATION NAME (LAST,FIRST, MI): DATE OF BIRTH (MM/DD/YY): HOSPITAL#: ACCESSION#: REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: SAMPLE INFORMATION CLINICAL INDICATION FETAL SPECIMEN TYPE Pregnancy at risk for specific genetic disorder DATE OF COLLECTION: (Complete FAMILIAL MUTATION information below) Amniotic Fluid: cc AMA PERFORMING PHYSICIAN: CVS: mg TA TC Abnormal Maternal Screen: Fetal Blood: cc GESTATIONAL AGE (GA) Calculation for AF-AFP* NTD TRI 21 TRI 18 Other: SELECT ONLY ONE: Abnormal NIPT (attach report): POC/Fetal Tissue, Type: TRI 21 TRI 13 TRI 18 Other: Cultured Amniocytes U/S DATE (MM/DD/YY): Abnormal U/S (SPECIFY): Cultured CVS GA ON U/S DATE: WKS DAYS PARENTAL BLOODS - REQUIRED FOR CMA -OR- Maternal Blood Date of Collection: Multiple Pregnancy Losses LMP DATE (MM/DD/YY): Parental Concern Paternal Blood Date of Collection: Other Indication (DETAIL AND ATTACH REPORT): *Important: U/S dating will be used if no selection is made. Name: Note: Results will differ depending on method checked. Last Name First Name U/S dating increases overall screening performance. Date of Birth: KNOWN FAMILIAL MUTATION/DISORDER SPECIFIC PRENATAL TESTING Notice: Prior to ordering testing for any of the disorders listed, you must call the lab and discuss the clinical history and sample requirements with a genetic counselor. -

Structural and Molecular Basis of the Assembly of the TRPP2/PKD1 Complex
Structural and molecular basis of the assembly of the TRPP2/PKD1 complex Yong Yua, Maximilian H. Ulbrichb, Ming-Hui Lia, Zafir Buraeia, Xing-Zhen Chend, Albert C. M. Onge, Liang Tonga, Ehud Y. Isacoffb,c, and Jian Yanga,1 aDepartment of Biological Sciences, Columbia University, New York, NY 10027; bDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; cMaterial Science and Physical Bioscience Divisions, Lawrence Berkeley National Laboratory, Berkeley, CA 94720; dMembrane Protein Research Group, Department of Physiology, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada T6G 2H7; and eKidney Genetics Group, Academic Unit of Nephrology, Sheffield Kidney Institute, University of Sheffield Medical School, Sheffield S10 2RX, United Kingdom Edited by Christopher Miller, Brandeis University, Waltham, MA, and approved May 20, 2009 (received for review April 2, 2009) Mutations in PKD1 and TRPP2 account for nearly all cases of autoso- structural basis of the homomeric assembly of a functionally mal dominant polycystic kidney disease (ADPKD). These 2 proteins important TRPP2 coiled-coil domain, uncovers the crucial role form a receptor/ion channel complex on the cell surface. Using a of this domain in the assembly of the TRPP2/PKD1 complex, and combination of biochemistry, crystallography, and a single-molecule reveals an unexpected subunit stoichiometry of this complex. method to determine the subunit composition of proteins in the plasma membrane of live cells, we find that this complex contains 3 Results TRPP2 and 1 PKD1. A newly identified coiled-coil domain in the C Subunit Stoichiometry of the TRPP2 Homomultimer Expressed in a Cell terminus of TRPP2 is critical for the formation of this complex. -

Original Article Identification of B Cells Participated in the Mechanism of Postmenopausal Women Osteoporosis Using Microarray Analysis
Int J Clin Exp Med 2015;8(1):1027-1034 www.ijcem.com /ISSN:1940-5901/IJCEM0003564 Original Article Identification of B cells participated in the mechanism of postmenopausal women osteoporosis using microarray analysis Bing Yan*, Jie Li*, Li Zhang Department of Orthopedics, Shandong Provincial Traditional Chinise Medical Hospital, Jinan 250014, Shandong Province, China. *Equal contributors. Received November 3, 2014; Accepted January 7, 2015; Epub January 15, 2015; Published January 30, 2015 Abstract: To further understand the molecular mechanism of lymphocytes B cells in postmenopausal women os- teoporosis. Microarray data (GSE7429) were downloaded from Gene Expression Omnibus, in which B cells were separated from the whole blood of postmenopausal women, including 10 with high bone mineral density (BMD) and 10 with low BMD. Differentially expressed genes (DEGs) between high and low BMD women were identified by Student’s t-test, and P < 0.01 was used as the significant criterion. Functional enrichment analysis was performed for up- and down-regulated DEGs using KEGG, REACTOME, and Gene Ontology (GO) databases. Protein-protein inter- action network (PPI) of up- and down-regulated DEGs was respectively constructed by Cytoscape software using the STRING data. Total of 169 up-regulated and 69 down-regulated DEGs were identified. Functional enrichment analy- sis indicated that the genes (ITPA, ATIC, UMPS, HPRT1, COX10 and COX15) might participate in metabolic pathways, MAP3K10 and MAP3K9 might participate in the activation of JNKK activity, COX10 and COX15 might involve in mitochondrial electron transport, and ATIC, UMPS and HPRT1 might involve in transferase activity. MAPK3, ITPA, ATIC, UMPS and HPRT1 with a higher degree in PPI network were identified. -

1 1 2 3 Cell Type-Specific Transcriptomics of Hypothalamic
1 2 3 4 Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to 5 weight-loss 6 7 Fredrick E. Henry1,†, Ken Sugino1,†, Adam Tozer2, Tiago Branco2, Scott M. Sternson1,* 8 9 1Janelia Research Campus, Howard Hughes Medical Institute, 19700 Helix Drive, Ashburn, VA 10 20147, USA. 11 2Division of Neurobiology, Medical Research Council Laboratory of Molecular Biology, 12 Cambridge CB2 0QH, UK 13 14 †Co-first author 15 *Correspondence to: [email protected] 16 Phone: 571-209-4103 17 18 Authors have no competing interests 19 1 20 Abstract 21 Molecular and cellular processes in neurons are critical for sensing and responding to energy 22 deficit states, such as during weight-loss. AGRP neurons are a key hypothalamic population 23 that is activated during energy deficit and increases appetite and weight-gain. Cell type-specific 24 transcriptomics can be used to identify pathways that counteract weight-loss, and here we 25 report high-quality gene expression profiles of AGRP neurons from well-fed and food-deprived 26 young adult mice. For comparison, we also analyzed POMC neurons, an intermingled 27 population that suppresses appetite and body weight. We find that AGRP neurons are 28 considerably more sensitive to energy deficit than POMC neurons. Furthermore, we identify cell 29 type-specific pathways involving endoplasmic reticulum-stress, circadian signaling, ion 30 channels, neuropeptides, and receptors. Combined with methods to validate and manipulate 31 these pathways, this resource greatly expands molecular insight into neuronal regulation of 32 body weight, and may be useful for devising therapeutic strategies for obesity and eating 33 disorders. -

Mechanistic Analysis of an Extracellular Signal-Regulated
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2020/10/26/jpet.120.000266.DC1 1521-0103/376/1/84–97$35.00 https://doi.org/10.1124/jpet.120.000266 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 376:84–97, January 2021 Copyright ª 2020 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. Mechanistic Analysis of an Extracellular Signal–Regulated Kinase 2–Interacting Compound that Inhibits Mutant BRAF-Expressing Melanoma Cells by Inducing Oxidative Stress s Ramon Martinez, III,1 Weiliang Huang,1 Ramin Samadani, Bryan Mackowiak, Garrick Centola, Lijia Chen, Ivie L. Conlon, Kellie Hom, Maureen A. Kane, Steven Fletcher, and Paul Shapiro Department of Pharmaceutical Sciences, University of Maryland, Baltimore- School of Pharmacy, Baltimore, Maryland Received August 3, 2020; accepted October 6, 2020 Downloaded from ABSTRACT Constitutively active extracellular signal–regulated kinase (ERK) 1/2 (MEK1/2) or ERK1/2. Like other ERK1/2 pathway inhibitors, 1/2 signaling promotes cancer cell proliferation and survival. We SF-3-030 induced reactive oxygen species (ROS) and genes previously described a class of compounds containing a 1,1- associated with oxidative stress, including nuclear factor ery- dioxido-2,5-dihydrothiophen-3-yl 4-benzenesulfonate scaffold throid 2–related factor 2 (NRF2). Whereas the addition of the jpet.aspetjournals.org that targeted ERK2 substrate docking sites and selectively ROS inhibitor N-acetyl cysteine reversed SF-3-030–induced inhibited ERK1/2-dependent functions, including activator ROS and inhibition of A375 cell proliferation, the addition of protein-1–mediated transcription and growth of cancer cells NRF2 inhibitors has little effect on cell proliferation. -

Universities of Leeds, Sheffield and York
promoting access to White Rose research papers Universities of Leeds, Sheffield and York http://eprints.whiterose.ac.uk/ This is an author produced version of a paper published in Proceedings of the National Academy of Sciences of the United States of America. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/9053 Published paper Yu, Y., Ulbrich, M., Li, M.H., Buraei, Z., Chen, X.Z., Ong, A.C.M., Tong, L., Isacoff, E.Y., Yang, J. (2009) Structural and molecular basis of the assembly of the TRPP2/PKD1 complex, Proceedings of the National Academy of Sciences of the United States of America, 106 (28), pp. 11558-11563 http://dx.doi.org/10.1073/pnas.0903684106 White Rose Research Online [email protected] 1 Classification-- BIOLOGICAL SCIENCES: Biochemistry Structural and molecular basis of the assembly of the TRPP2/PKD1 complex Yong Yua, Maximilian H. Ulbrichb, Ming-hui Lia, Zafir Buraeia, Xing-Zhen Chend, Albert C.M. Onge, Liang Tonga, Ehud Y. Isacoffb,c and Jian Yanga, 1 aDepartment of Biological Sciences, Columbia University, New York, NY 10027, USA bDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA cMaterial Sciences & Physics Biosciences Divisions, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA dMembrane Protein Research Group, Department of Physiology, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, Alberta, T6G 2H7, Canada eKidney Genetics Group, Academic Unit of Nephrology, Sheffield Kidney Institute, School of Medicine and Biomedical Sciences, University of Sheffield, Sheffield, UK 2 1 To whom correspondence should be addressed. Jian Yang Department of Biological Sciences, 917 Fairchild Center, MC2462, Columbia University, New York, NY 10027 Phone: (212)-854-6161; Fax: (212)-531-0425 Email: [email protected] Author contributions: Y.Y., M.H.U., M.L., Z.B., X-Z.C., A.C.M.O, L.T., E.Y.I., and J.Y. -
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -
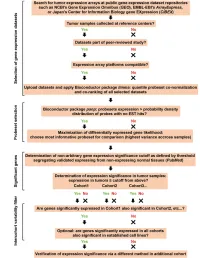
Supplementary Data
SUPPLEMENTAL INFORMATION A study restricted to chemokine receptors as well as a genome-wide transcript analysis uncovered CXCR4 as preferentially expressed in Ewing's sarcoma (Ewing's sarcoma) cells of metastatic origin (Figure 4). Transcriptome analyses showed that in addition to CXCR4, genes known to support cell motility and invasion topped the list of genes preferentially expressed in metastasis-derived cells (Figure 4D). These included kynurenine 3-monooxygenase (KMO), galectin-1 (LGALS1), gastrin-releasing peptide (GRP), procollagen C-endopeptidase enhancer (PCOLCE), and ephrin receptor B (EPHB3). KMO, a key enzyme of tryptophan catabolism, has not been linked to metastasis. Tryptophan and its catabolites, however, are involved in immune evasion by tumors, a process that can assist in tumor progression and metastasis (1). LGALS1, GRP, PCOLCE and EPHB3 have been linked to tumor progression and metastasis of several cancers (2-4). Top genes preferentially expressed in L-EDCL included genes that suppress cell motility and/or potentiate cell adhesion such as plakophilin 1 (PKP1), neuropeptide Y (NPY), or the metastasis suppressor TXNIP (5-7) (Figure 4D). Overall, L-EDCL were enriched in gene sets geared at optimizing nutrient transport and usage (Figure 4D; Supplementary Table 3), a state that may support the early stages of tumor growth. Once tumor growth outpaces nutrient and oxygen supplies, gene expression programs are usually switched to hypoxic response and neoangiogenesis, which ultimately lead to tumor egress and metastasis. Accordingly, gene sets involved in extracellular matrix remodeling, MAPK signaling, and response to hypoxia were up-regulated in M-EDCL (Figure 4D; Supplementary Table 4), consistent with their association to metastasis in other cancers (8, 9).