Development and Application of a Next-Generation- Sequencing (NGS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Shimada 2017.Pdf
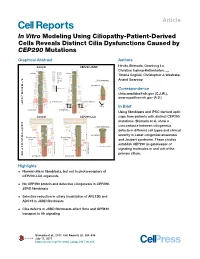
Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Graphical Abstract Authors Control CEP290-JSRD Hiroko Shimada, Quanlong Lu, Christine Insinna-Kettenhofen, ..., Cilium GPR161 Cilium ADCY3 Tiziana Cogliati, Christopher J. Westlake, Smo ARL13B F CEP290 Cell membrane Anand Swaroop I Gli2 B Cytoplasm R Other proteins O Correspondence Axoneme B Axoneme [email protected] (C.J.W.), L A [email protected] (A.S.) S Cell membrane Cell membrane T Vesicles In Brief Cytoplasm Cytoplasm Using fibroblasts and iPSC-derived optic Control CEP290-LCA cups from patients with distinct CEP290 mutations, Shimada et al. show a P H Cell membrane concordance between ciliogenesis O T ? defects in different cell types and clinical Axoneme ? O ? Axoneme ? Cytoplasm severity in Leber congenital amaurosis R ? Connecting Connecting ? E Cilium Cilium ? and Joubert syndrome. These studies Cell membrane C Cell membrane E establish CEP290 as gatekeeper of P Docked mother centriole T ?? ? ? signaling molecules in and out of the O primary cilium. R Cytoplasm Cytoplasm Highlights d Normal cilia in fibroblasts, but not in photoreceptors of CEP290-LCA organoids d No CEP290 protein and defective ciliogenesis in CEP290- JSRD fibroblasts d Selective reduction in ciliary localization of ARL13B and ADCY3 in JSRD fibroblasts d Cilia defects in JSRD fibroblasts affect Smo and GPR161 transport in Hh signaling Shimada et al., 2017, Cell Reports 20, 384–396 July 11, 2017 http://dx.doi.org/10.1016/j.celrep.2017.06.045 Cell Reports Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Hiroko Shimada,1 Quanlong Lu,2 Christine Insinna-Kettenhofen,2 Kunio Nagashima,3 Milton A. -

Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Journal of Pediatric Genetics 3 (2014) 65–78 65 DOI 10.3233/PGE-14090 IOS Press Unraveling the genetics of Joubert and Meckel-Gruber syndromes Katarzyna Szymanska, Verity L. Hartill and Colin A. Johnson∗ Department of Ophthalmology and Neuroscience, University of Leeds, Leeds, UK Received 27 May 2014 Revised 11 July 2014 Accepted 14 July 2014 Abstract. Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are recessive neurodevelopmental conditions caused by mutations in proteins that are structural or functional components of the primary cilium. In this review, we provide an overview of their clinical diagnosis, management and molecular genetics. Both have variable phenotypes, extreme genetic heterogeneity, and display allelism both with each other and other ciliopathies. Recent advances in genetic technology have significantly improved diagnosis and clinical management of ciliopathy patients, with the delineation of some general genotype-phenotype correlations. We highlight those that are most relevant for clinical practice, including the correlation between TMEM67 mutations and the JBTS variant phenotype of COACH syndrome. The subcellular localization of the known MKS and JBTS proteins is now well-described, and we discuss some of the contemporary ideas about ciliopathy disease pathogenesis. Most JBTS and MKS proteins localize to a discrete ciliary compartment called the transition zone, and act as structural components of the so-called “ciliary gate” to regulate the ciliary trafficking of cargo proteins or lipids. Cargo proteins include enzymes and transmembrane proteins that mediate intracellular signaling. The disruption of transition zone function may contribute to the ciliopathy phenotype by altering the composition of the ciliary membrane or axoneme, with impacts on essential developmental signaling including the Wnt and Shh pathways as well as the regulation of secondary messengers such as inositol-1,4,5-trisphosphate (InsP3) and cyclic adenosine monophosphate (cAMP). -

The Mineralocorticoid Receptor Leads to Increased Expression of EGFR
www.nature.com/scientificreports OPEN The mineralocorticoid receptor leads to increased expression of EGFR and T‑type calcium channels that support HL‑1 cell hypertrophy Katharina Stroedecke1,2, Sandra Meinel1,2, Fritz Markwardt1, Udo Kloeckner1, Nicole Straetz1, Katja Quarch1, Barbara Schreier1, Michael Kopf1, Michael Gekle1 & Claudia Grossmann1* The EGF receptor (EGFR) has been extensively studied in tumor biology and recently a role in cardiovascular pathophysiology was suggested. The mineralocorticoid receptor (MR) is an important efector of the renin–angiotensin–aldosterone‑system and elicits pathophysiological efects in the cardiovascular system; however, the underlying molecular mechanisms are unclear. Our aim was to investigate the importance of EGFR for MR‑mediated cardiovascular pathophysiology because MR is known to induce EGFR expression. We identifed a SNP within the EGFR promoter that modulates MR‑induced EGFR expression. In RNA‑sequencing and qPCR experiments in heart tissue of EGFR KO and WT mice, changes in EGFR abundance led to diferential expression of cardiac ion channels, especially of the T‑type calcium channel CACNA1H. Accordingly, CACNA1H expression was increased in WT mice after in vivo MR activation by aldosterone but not in respective EGFR KO mice. Aldosterone‑ and EGF‑responsiveness of CACNA1H expression was confrmed in HL‑1 cells by Western blot and by measuring peak current density of T‑type calcium channels. Aldosterone‑induced CACNA1H protein expression could be abrogated by the EGFR inhibitor AG1478. Furthermore, inhibition of T‑type calcium channels with mibefradil or ML218 reduced diameter, volume and BNP levels in HL‑1 cells. In conclusion the MR regulates EGFR and CACNA1H expression, which has an efect on HL‑1 cell diameter, and the extent of this regulation seems to depend on the SNP‑216 (G/T) genotype. -

Prenatal Exome Sequencing in Anomalous Fetuses: New Opportunities and Challenges
© American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges Neeta L. Vora, MD1, Bradford Powell, MD, PhD2, Alicia Brandt, MS2, Natasha Strande, PhD3,4, Emily Hardisty, MS, CGC1, Kelly Gilmore, MS, CGC1, Ann Katherine M. Foreman, MS, CGC2,5, Kirk Wilhelmsen, MD, PhD6, Chris Bizon, PhD6, Jason Reilly, BA6, Phil Owen, BS6, Cynthia M. Powell, MD, MS2,7, Debra Skinner, PhD, MA8, Christine Rini, PhD9, Anne D. Lyerly, MD, MA10, Kim A. Boggess, MD1, Karen Weck, MD3,4, Jonathan S. Berg, MD, PhD2 and James P. Evans, MD, PhD2,10 Purpose: We investigated the diagnostic and clinical performance the following genes: COL1A1, MUSK, KCTD1, RTTN, TMEM67, of exome sequencing in fetuses with sonographic abnormalities PIEZO1 and DYNC2H1. One additional case revealed a de novo with normal karyotype and microarray and, in some cases, normal nonsense mutation in a novel candidate gene (MAP4K4). gene-specific sequencing. The perceived likelihood that exome sequencing would explain Methods: Exome sequencing was performed on DNA from 15 the results (5.2 on a 10-point scale) was higher than the anomalous fetuses and from the peripheral blood of their approximately 30% diagnostic yield discussed in pretest counseling. parents. Parents provided consent to be informed of diagnostic Conclusion: Exome sequencing had diagnostic utility in a highly results in the fetus, medically actionable findings in the parents, select population of fetuses where a genetic diagnosis was highly and their identification as carrier couples for significant auto- suspected. Challenges related to genetics literacy and variant somal recessive conditions. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

Yan Et Al. Supplementary Material
SUPPLEMENTARY MATERIAL FOR: CELL ATLAS OF THE HUMAN FOVEA AND PERIPHERAL RETINA Wenjun Yan*, Yi-Rong Peng*, Tavé van Zyl*, Aviv Regev, Karthik Shekhar, Dejan Juric, and Joshua R, Sanes^ *Co-First authors ^Author for correspondence, [email protected] Figure S1 tSNE visualization showing contributions to cell types by batch for photoreceptors (a), horizontal cells (b), bipolar cells (c), amacrine cells (d), retinal ganglion cells (e) and non-neuronal cells (f). Each dot represents one cell. Colors distinguish retina samples. Source of each sample is shown in Table S1. Overall, batch eFFects were minimal. Figure S2 Violin and superimposed box plots showing expression of OPN4 in RGC clusters Figure S3 Heat maps showing expression patterns of disease genes by cell classes in the Fovea and periphery. Only genes expressed by more than 20% of cells in any individual class in either Fovea or peripheral cells are plotted. Table S1 Information on donors from whom retinal cells were obtained for scRNA-seq proFiling. Table S2 Publications reporting single cell or single nucleus profiling on cells from human retina. 1 Figure S1 a b c PR HC BP H1 H2F1 H2F2 H3 tSNE1 tSNE1 H4 tSNE1 H5 H9 H11 tSNE2 tSNE2 tSNE2 e AC f RGC g Non-neuronal tSNE1 tSNE1 tSNE1 tSNE2 tSNE2 tSNE2 Figure S2 OPN4 4 2 log Expression 0 MG-ON MG-OFF PG-OFF PG-ON hRGC5 hRGC6 hRGC7 hRGC8 hRGC9 hRGC10 hRGC11 hRGC12 Figure S3 Fovea Fovea Peripheral Peripheral Rods Cones BP HC AC RGC Muller Astro MicG Endo Rods Cones BP HC AC RGC Muller Astro MicG Endo ARL13B MAPK8IP3 EXOC6 LSM4 Expression -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

An Advance About the Genetic Causes of Epilepsy
E3S Web of Conferences 271, 03068 (2021) https://doi.org/10.1051/e3sconf/202127103068 ICEPE 2021 An advance about the genetic causes of epilepsy Yu Sun1, a, *, †, Licheng Lu2, b, *, †, Lanxin Li3, c, *, †, Jingbo Wang4, d, *, † 1The School of Molecular and Cellular Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801-3633, US 2High School Affiliated to Shanghai Jiao Tong University, Shanghai, 200441, China 3Applied Biology program, University of British Columbia, Vancouver, V6r3b1, Canada 4School of Chemical Machinery and Safety, Dalian University of Technology, Dalian, 116023, China †These authors contributed equally. Abstract: Human hereditary epilepsy has been found related to ion channel mutations in voltage-gated channels (Na+, K+, Ca2+, Cl-), ligand gated channels (GABA receptors), and G-protein coupled receptors, such as Mass1. In addition, some transmembrane proteins or receptor genes, including PRRT2 and nAChR, and glucose transporter genes, such as GLUT1 and SLC2A1, are also about the onset of epilepsy. The discovery of these genetic defects has contributed greatly to our understanding of the pathology of epilepsy. This review focuses on introducing and summarizing epilepsy-associated genes and related findings in recent decades, pointing out related mutant genes that need to be further studied in the future. 1 Introduction Epilepsy is a neurological disorder characterized by 2 Malfunction of Ion channel epileptic seizures caused by abnormal brain activity. 1 in Functional variation in voltage or ligand-gated ion 100 (50 million people) people are affected by symptoms channel mutations is a major cause of idiopathic epilepsy, of this disorder worldwide, with men, young children, and especially in rare genetic forms. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9