RECOVERY Protocol V3.0 (2020-04-07)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old Road Campus
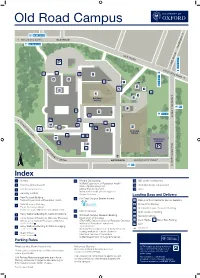
Old Road Campus 4a, 4b, 4c, U5 n o t Oxford City Centre g OLD ROAD n i d 4a, 4b, 4c, U5 a e H K O L L A D R O W AD E M I L 4,4a,4b,4c, U1X,U5 A41 42 4,4a,4b,4c,U5 Rin D 4 g R oad 6 7 1 13 3 11 C H U R C H I L L 900 D 2 R I 700, 900 V E E O x f o 5 A C rd 12 C i ty 10 C e n t re B 8 CAR PARK C 9 h u r c R F h i O l l O S H E o V s N E p L i T t DRI a VE ENTRANCE ROOSEVELT DRIVE l 900, ST2 Index 1 The Triangle Nursery 9 Old Road Campus Estates Annexe 13 Boundary Brook House Interserve Joint Research Office Kennedy Institute 2 - Research Services, Medical Sciences Division Old Road Campus Research Building 10 - Clinical Trials and Research Governance 3 New Richards Building Department of Oncology - Human Tissue Governance CRUK/MRC Oxford Institute for Radiation Oncology - Medical Sciences Division Business Development 4 NDM Research Building Institute of Biomedical Engineering Nuffield Department of Primary Care Health Sciences Target Discovery Institute Jenner Institute Medical Sciences Divisional Safety Officers Centre for Tropical Medicine and Global Health Bodleian Knowledge Centre (Library Services) Medical Sciences Division IT Services 5 Wellcome Centre for Human Genetics (WHG) Ludwig Institute for Cancer Research Structural Genomics Consortium 6 Henry Wellcome Building for Molecular Physiology Nuffield Department of Surgical Sciences Loading Bays and Delivery Offices of the Nuffield Professor of Medicine ENTRANCE VIA BUILDING 5 11 Big Data Institute A Wellcome Trust Centre for Human Genetics 7 Henry Wellcome Building for Particle Imaging -

Downloaded From
A Thesis Submitted for the Degree of PhD at the University of Warwick Permanent WRAP URL: http://wrap.warwick.ac.uk/149337 Copyright and reuse: This thesis is made available online and is protected by original copyright. Please scroll down to view the document itself. Please refer to the repository record for this item for information to help you to cite it. Our policy information is available from the repository home page. For more information, please contact the WRAP Team at: [email protected] warwick.ac.uk/lib-publications A Randomized Trial of Neprilysin Inhibition with Sacubitril/valsartan vs Irbesartan in Chronic Kidney Disease by Dr Parminder Kaur Judge Thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy (PhD) in Medicine University of Warwick & Medical Research Council Population Health Research Unit, Nuffield Department of Population Health, University of Oxford Submitted June 2020 1 Contents Section Page List of Tables 7 List of Figures 9 Acknowledgements 10 Declarations 12 Inclusion of Published Work 13 Abstract 14 Chapters 1 List of abbreviations 15 2 Introduction 19 3 Natriuretic peptide system and neprilysin 35 2 4 Angiotensin receptor-neprilysin inhibitor (ARNI) 46 Effects on renal function 50 Effects on albuminuria 53 5 Methods 65 Trial organisation 70 Staff training 70 Data management 70 Trial treatments 70 Consent 72 Biological samples 73 Randomized treatment and blinding 76 Blood and urine samples 76 3 Adverse events and compliance with trial treatment 77 Physical measurements -

1 RECOVERY Central Coordinating Office Nuffield Department Of
RECOVERY Central Coordinating Office Nuffield Department of Population Health Richard Doll Building Old Road Campus Roosevelt Drive Oxford OX3 7LF 24th May 2020 To all RECOVERY Principal Investigators Recruitment to the RECOVERY trial (including the Hydroxychloroquine arm) REMAINS OPEN On Friday 22nd May we received a letter from the MHRA in which they notified us of their concerns relating to the use of hydroxychloroquine as a treatment for patients with COVID-19 in the light of the recent publication by Mehra et al in The Lancet on 22 May 2020. We have held two videoconferences with the MHRA and provided a detailed response which is summarised below. This morning we have received written confirmation from the MHRA that, “it is acceptable to allow continued randomisation into the hydroxychloroquine arm of the trial.” Further details are provided below: • There have been two recent observational analyses of the use of hydroxychloroquine (HCQ) for treatment of COVID-19. One reported no significant effect on the outcome of intubation or death (Geleris et al, New Engl J Med 7 May 2020), while the other reported a hazard ratio of 1.33 for all-cause mortality (Mehra et al, Lancet 22 May 2020). The authors of both papers conclude that their findings should be interpreted with caution, are not definitive, and that randomised controlled trials are required to reach any conclusion about the benefit or harm of HCQ for COVID-19. • The RECOVERY trial is currently the largest randomised controlled trial of HCQ for COVID-19 but is not yet sufficiently large to detect (or rule out) moderate yet important treatment effects. -

Nuffield Department of Population Health
Nuffield Department of Population Health Richard Doll Building, Old Road Campus, Headington, Oxford OX3 7LF Tel: +44 (0)1865 743743, Fax: +44 (0)1865 743985, email: [email protected] _______________________________________________________________________________________________ 14 April 2014 Dr Fiona Godlee Editor, BMJ BMJ Publishing Group Tavistock Square London WC1H 9JR Dear Fiona NOT FOR PUBLICATION Thank you for responding so rapidly to my letter of 31 March, but unfortunately the key points that I raised have not been addressed properly either by the letters from Abramson et al and Malhotra or by your response of 1 April. I can assure you that this issue is not at all personal. As indicated previously, it seems highly likely that the misleading claims that have been reported by the BMJ about side- effect rates with statins will lead to large numbers of unnecessary heart attacks, strokes and premature deaths because patients at elevated risk will be dissuaded from taking statins. My concern is with the BMJ’s failure to correct promptly and prominently such a serious mistake when it has major public health implications. As I explained to you when we met, the evidence cited by Abramson et al and Malhotra does not support their claims that statins cause side-effects in 18-20% of patients. For the sake of clarity, a “statin-related adverse event” (which is what was studied in the paper by Zhang et al that is being cited) is not necessarily caused by, or a side-effect of, a statin. Consequently, it is a serious misrepresentation of the evidence for Abramson et al and Malhotra to state that it is. -

Job Description and Selection Criteria Introduction
George Institute for Global Health at University of Oxford University of Oxford New Richards Building Old Road Campus, Roosevelt Drive Oxford OX3 7LF United Kingdom T: +44 1865 617 203 F: +44 1865 617 202 [email protected] www.georgecentre.ox.ac.uk Job description and selection criteria Job title Executive Director, The George Institute for Global Health, China and James Martin Fellow, The George Institute for Global Health, University of Oxford Division Medical Sciences Department Nuffield Department of Population Health Location The George Institute at the University of Oxford, Richard Doll Building, Old Road Campus, Headington, Oxford (30%) and The George Institute at Peking University Health Science Center, Haidian District, Beijing (70%) Grade and salary Competitive Salary (RSIV grade) Hours Full time Contract type Fixed term, 5 Years Reporting to Principal Director, The George Institute for Global Health Vacancy reference Introduction The University of Oxford is a complex and stimulating organisation, which enjoys an international reputation as a world-class centre of excellence in research and teaching. It employs over 10,000 staff and has a student population of over 20,000. Most staff are directly appointed and managed by one of the University’s 130 departments or other units within a highly devolved operational structure - this includes 5,600 ‘academic- related’ staff (postgraduate research, computing, senior library, and administrative staff) and 2,700 ‘support’ staff (including clerical, library, technical, and manual staff). There are also over 1,600 academic staff (professors, readers, lecturers), whose appointments are in the main overseen by a combination of broader divisional and local faculty board/departmental structures. -
Old Road Campus
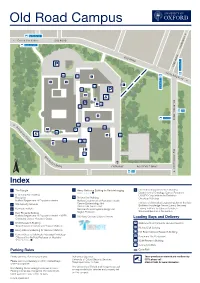
Old Road Campus 4a, 4b, 4c, U5 n Oxford City Centre OLD ROAD gto din 4a, 4b, 4c, U5 a He K O L LD A ROAD 4 W 4a E M 4b I 4c L U1X U5 A 414 2 Ri D 6 ng Ro □ 4 ad 4a 8 ■9 4b 1 4c U5 5 2 10 15 CHURCHILL DRIVE ■ 3 • BUILDING 13 WORKS 900 17 4 700, 900 E O x f or 7 A C d 16 C ity ■ Cent 14 BUILDING WORKS re • B 11 TEMPORARY CARPARK Churchill Hospital 12 [eJ R O O S E N V EL T DRIVE ENTRANCE ROOSEVELT DRIVE 900 Index 1 Nursery 11 Richard Doll Building 15 BDI (under construction) ■ Nuffi eld Department of Population Health ■ Kitchens (deliveries only) ■ Amenities (under construction) ■2 Cancer Epidemiology Unit ■16 3 Site Security Services Clinical Trial Service Unit 17 Mace National Perinatal Epidemiology Unit ■ ■ Kennedy Institute 4 Regius Professor Loading Bays and Delivery ■ New Richards Building 5 12 Old Road Campus Estates Annexe ■ Nuffi eld Department of Population Health ■ ~4'- □A Wellcome Trust Centre for Human Genetics NDM Research Building B Richard Doll Building 6 'j Interserve Ingenuity at work □ ■ Target Discovery Institute C Old Road Campus Research Building Centre for Tropical Medicine and Global Health □ 13 MSD IT Services D NDM Research Building 7 Henry Wellcome Building for Genomic Medicine ■ □ ■ 14 Old Road Campus Research Building E Kennedy Institute 8 Henry Wellcome Building for Molecular Physiology ■ Department of Oncology □ ■ Offi ces of the Nuffi eld Professor of Medicine CRUK/MRC Oxford Institute for Radiation Oncology ~ Cycle Racks ~ Motor Bike Parking ENTRANCE VIA BUILDING ■7 Institute of Biomedical Engineering [Ii!] Café 9 Henry Wellcome Building for Particle Imaging Jenner Institute ~ Entrances ■ ENTRANCE VIA BUILDING ■7 Bodleian Knowledge Centre (Library Services) Ludwig Institute for Cancer Research Quad Offices 10 Structural Genomics Consortium ENTRANCE VIA BUILDING 7 ■ ■ Nuffi eld Department of Surgical Sciences Parking Rules Private parking. -

Health Services Research Unit
Participant Information Sheet (version 26.6.2018) Health Services Research Unit Nuffield Department of Population Health Richard Doll Building, Old Road Campus, Headington, Oxford OX3 7LF Telephone 01865 289432 or email [email protected] Telephone 01865 289424 or email [email protected] Pretesting new items for the Parkinson’s Disease Questionnaire (PDQ) (ETHICS APPROVAL REFERENCE R58649/RE001) 1. What is the purpose of the study? The Parkinson’s Disease Questionnaire (PDQ) was developed in 1997 to measure the effects of Parkinson’s on every day life. Researchers in the Health Services Research Unit (HSRU) at the University of Oxford have recently conducted a series of interviews with people with Parkinson’s in order to explore whether additional questions for the PDQ are required in light of changes in the management of Parkinson’s over the last 20 years. Between 1 August and 30 October 2018 we will be conducting interviews with a small number of people with Parkinson’s to test a pool of newly generated PDQ questions. This is to ensure that the proposed questions are relevant, acceptable, unambiguous and usable. 2. Why have I been asked to take part in the study? You are being approached because you are a member of Parkinson’s UK and your local branch or the Research Support Network is helping to promote the project. We are looking for between 12 and 20 adults with Parkinson’s to take part. You must be over 18 years old, live in the UK, have access to the internet and use your computer or other electronic device to complete the proposed new questions online. -
Parking Rules Loading Bays and Delivery
Old Road Campus 4a, 4b, 4c, U5 n o Oxford City Centre OLD ROAD t ng 4a, 4b, 4c, U5 di ea H K O L LD A R 4 OA W D E 4a M 4b I L 4c U1X U5 A4 142 Rin E 6 g Ro 4 ad 4a 8 9 4b 1 4c U5 5 2 CHURCHILL DRIVE 3 D 900 4 700, 900 F O x for 7 A C d Cit y 12 C ent re B 10 11 Churchill Hospital R O O S E N V EL T D RIVE ENTRANCE ROOSEVELT DRIVE 900 Index 1 The Triangle 9 Henry Wellcome Building for Particle Imaging 12 Old Road Campus Research Building ENTRANCE VIA BUILDING 7 Department of Oncology, Cancer Research 2 Rosemary Rue Building UK/MRC Gray Institute for Radiation 10 Reception Richard Doll Building Oncology & Biology Nuffield Department of Population Health Nuffield Department of Population Health Institute of Biomedical Engineering Jenner Institute 3 Site Security Services Cancer Epidemiology Unit Clinical Trial Service Unit Bodleian Knowledge Centre (Library Services) 4 Kennedy Institute National Perinatal Epidemiology Unit Ludwig Institute for Cancer Research Structural Genomics Consortium 5 New Richards Building Regius Professor Nuffield Department of Population Health – NPEU 11 Old Road Campus Estates Annexe Childhood Cancer Research Group Loading Bays and Delivery 6 NDM Research Building A Wellcome Trust Centre for Human Genetics Target Discovery Institute and Tropical Medicine B Richard Doll Building 7 Henry Wellcome Building for Genomic Medicine C Old Road Campus Research Building 8 Henry Wellcome Building for Molecular Physiology D Offices of the Nuffield Professor of Medicine Rosemary Rue Restaurant ENTRANCE VIA BUILDING 7 NDM Research Building Kennedy Institute Parking Rules Cycle Path Private parking. -

Aspirin in Patients Admitted to Hospital with COVID-19 (RECOVERY): a Randomised, Controlled, Open-Label, Platform Trial
medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258132; this version posted June 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . Aspirin for COVID-19 1 2 3 Aspirin in patients admitted to hospital with 4 COVID-19 (RECOVERY): a randomised, controlled, 5 open-label, platform trial 6 7 Running title: Aspirin for COVID-19 8 9 RECOVERY Collaborative Group* 10 11 12 *The writing committee and trial steering committee are listed at the end of this 13 manuscript and a complete list of collaborators in the Randomised Evaluation of 14 COVID-19 Therapy (RECOVERY) trial is provided in the Supplementary Appendix. 15 16 Correspondence to: Prof Peter W Horby and Prof Martin J Landray, RECOVERY Central 17 Coordinating Office, Richard Doll Building, Old Road Campus, Roosevelt Drive, Oxford OX3 18 7LF, United Kingdom. 19 Email: [email protected] 20 21 Word count: 22 Abstract – 301 words 23 Main text – 3085 words 24 References – 33 25 Tables & Figures – 2 + 3 26 27 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258132; this version posted June 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

Active Monitoring for Atrial Fibrillation a Randomized Controlled Trial of Screening for Silent Atrial Fibrillation in H
Date and version No: 2021-06-08 Version 2.0 ISRCTN 15544176 AMALFI: Active Monitoring for AtriaL Fibrillation A randomized controlled trial of screening for silent atrial fibrillation in high-risk individuals. Ethics Ref: 19/LO/0220 Date and Version No: 2021-06-08 V2.0 Chief Investigator: Professor Louise Bowman Investigators: Professor Barbara Casadei Dr Guilherme Pessoa-Amorim Dr Nicholas Jones Dr Christine A’Court Mr Richard Bulbulia Mrs Georgina Buck Sponsor: University of Oxford Funder: NIHR, Oxford Biomedical Research Centre (BRC) Chief Investigator Signature: AMALFI Central Coordinating Office, Clinical Trial Service Unit, Richard Doll Building, Old Road Campus, Roosevelt Drive, Oxford OX3 7LF, UK Tel: Freephone 0808 164 5080 or: +44 (0) 1865 743884 Email: [email protected], website: www.amalfitrial.org Chief Investigator: Professor Louise Bowman Short Title: AMALFI Ethics Ref: 19/LO/0220 Page 1 of 26 Date and version No: 2021-06-08 Version 2.0 ISRCTN 15544176 TABLE OF CONTENTS 1. SYNOPSIS ............................................................................................................................................... 4 2. ABBREVIATIONS ..................................................................................................................................... 5 3. BACKGROUND AND RATIONALE ............................................................................................................ 6 3.1 AF: A global health problem associated with substantial morbidity and mortality ........................... -

Health Services Research Unit
Participant information sheet v 15.5.2018 Health Services Research Unit Nuffield Department of Population Health Richard Doll Building, Old Road Campus, Headington, Oxford OX3 7LF Telephone 01865 289424 or email [email protected] Telephone 01865 289432 or email [email protected] Pretesting the PDQ-Carer in electronic format Ethics Approval Reference: RS56991/REE001 1. What is the purpose of this research? Researchers in the Health Services Research Unit (HSRU) at the University of Oxford have recently developed the Parkinson’s Disease Carer Questionnaire (PDQ-Carer) in order to measure the effects of being a carer for someone with Parkinson’s disease. The PDQ-Carer is being transferred to an electronic format so it can be used on personal computers, laptops, tablet computers and smart phones. We are conducting interviews with a small number of carers for people with Parkinson’s disease to assess its usability and acceptability, to ensure that it can be used successfully online. 2. Why have I been invited to take part? You are being approached because you are an informal carer for someone with Parkinson’s who is a member of Parkinson’s UK and your local branch or the Research Support Network is helping to promote the project. For this research what is meant by ‘informal carer’ is a family member or friend who provides help and support to a person with Parkinson’s disease but is not paid for it. We are looking for between six and ten informal carers to complete an online questionnaire and be interviewed about it. -

Lectures and Seminars, Michaelmas Term 2015
WEDNESDay 7 octobEr 2015 • SUPPLEMENt (1) to No 5107 • VoL 146 Gazette Supplement Lectures and Seminars, Michaelmas term 2015 Witness Seminar (the UN orthopaedics, rheumatology and Islamic Studies at 70) 26 Musculoskeletal Sciences Reuters Institute for the Study of Pathology Journalism Pharmacology Reuters Institute for the Study of Charles Simonyi Lecture 26 Physiology, anatomy and Genetics Journalism/Nuffield Population Health Latin american centre Humanities 26 Psychiatry Foundation for Law, Justice and Society/ centre for Socio-Legal Studies TORCH | The Oxford Research Centre in Social Sciences 34 Learning Institute the Humanities Maison Française Rothermere American Institute anthropology and Museum Ethnography oxford Martin School Classics Archaeology Population ageing English Language and Literature Saïd Business School English/History/History of Art/Theology/ Education Colleges, Halls and Societies 44 Music Geography and the Environment all Souls/Magdalen History Intellectual Property research centre Balliol History/Politics and International Interdisciplinary Area Studies Relations Green templeton International Development (Queen Linguistics, Philology and Phonetics Elizabeth House) Keble Linguistics, Philology and Phonetics/ Law Lady Margaret Hall Medieval and Modern Languages Politics and International relations Magdalen Medieval and Modern Languages Social Policy and Intervention Mansfield Music Socio-legal Studies Nuffield Oriental Studies Sociology Oriel St Anne’s Mathematical, Physical and Department for Continuing St