Preparation and Reactions of Some Lower Tungsten Halides and Halide Complexes Theodore Martin Brown Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Geoffrey Wilkinson
THE LONG SEARCH FOR STABLE TRANSITION METAL ALKYLS Nobel Lecture, December 11, 1973 by G EOFFREY W ILKINSON Imperial College of Science & Technology, London, England Chemical compounds in which there is a single bond between a saturated car- bon atom and a transition metal atom are of unusual importance. Quite aside from the significance and role in Nature of the cobalt to carbon bonds in the vitamin B 12 system and possible metal to carbon bonds in other biological systems, we need only consider that during the time taken to deliver this lec- ture, many thousands, if not tens of thousands of tons of chemical compounds are being transformed or synthesised industrially in processes which at some stage involve a transition metal to carbon bond. The nonchemist will pro- bably be most familiar with polyethylene or polypropylene in the form of do- mestic utensils, packaging materials, children’s toys and so on. These materials are made by Ziegler-Natta* or Philipps’ catalysis using titanium and chro- mium respectively. However, transition metal compounds are used as catalysts in the synthesis of synthetic rubbers and other polymers, and of a variety of simple compounds used as industrial solvents or intermediates. For example alcohols are made from olefins, carbon monoxide and hydrogen by use of cobalt or rhodium catalysts, acetic acid is made by carbonylation of methanol using rhodium catalysts and acrylonitrile is dimerised to adiponitrile (for nylon) by nickel catalysts. We should also not forget that the huge quantities of petroleum hydrocarbons processed by the oil and petrochemical industry are re-formed over platinum, platinum-rhenium or platinum-germanium sup- ported on alumina. -

Influence of Water on Conductivity of Aluminium Halide Solutions in Acetonitrile
Influence of water on conductivity of aluminium halide solutions in acetonitrile L. LUX Department of Chemistry, Faculty of Metallurgy, Technical University, CS-043 85 Košice Received 7 September 1981 Accepted for publication 8 February 1982 Equilibria of the substitution and association reactions of AICI3 and AlBr3 with acetonitrile were investigated by measuring the conductivity of solutions at varying concentrations of A1C13, AlBr3, and water in the acetonitrile solution. Both halides behave in acetonitrile in the absence of water as relatively strong electrolytes. It has been revealed that water displaces acetoni trile from the solvate sphere of cation of both halides. As for AlBr3, the bromide ion is also displaced from the coordination sphere of aluminium and replaced by a neutral water molecule, which simultaneously results in an increased number of ionized particles in solution. The hydrate A1X3 -6H20 was identified as final product which separates from the solution by adding water. Изучены равновесия реакций замещения и ассоциации А1С13 и А1Вг3 с ацетонитрилом посредством измерения электропроводности раствора в зависимости от концентрации А1С13, А1Вг3, как и в зависимости от концентрации воды в растворе ацетонитрила. Оба галогенида ведут себя в ацетонитриле как сравнительно сильные электролиты в отсутствии воды. Обнаружено, что вода вытесняет ацетонитрил из сольватной обо лочки катиона у обоих галогенидов. У А1Вг3 доходит и к вытеснению иона брома из координационной сферы алюминия и его замещению нейтраль ной молекулой воды, что одновременно приводит к увеличению числа ионизированных частиц в растворе. Конечным продуктом, выделенным из раствора при добавлении воды был идентифицированный гидрат А1Х3-6Н20. Acetonitrile is a typical aprotic solvent which is widely used in electrochemical research because of its profitable properties, i.e. -

Molecular Dynamics Simulation of Ion Mobility. 2. Alkali Metal and Halide Ions Using the SPC/E Model for Water at 25 °C†
+ + 1420 J. Phys. Chem. 1996, 100, 1420-1425 Molecular Dynamics Simulation of Ion Mobility. 2. Alkali Metal and Halide Ions Using the SPC/E Model for Water at 25 °C† Song Hi Lee Department of Chemistry, Kyungsung UniVersity, Pusan 608-736, Korea Jayendran C. Rasaiah* Department of Chemistry, UniVersity of Maine, Orono, Maine 04469 ReceiVed: October 16, 1995; In Final Form: NoVember 10, 1995X We present results of computer simulations of the mobilities of the alkali metal ions (Li+, Na+,K+, Rb+, and Cs+) and the halides (F-, Cl-, Br-, and I-) at 25 °C using the SPC/E model for water and ion-water parameters fitted to the binding energies of small clusters of ions. A simple truncation of the ion-water and water- water potentials was used, and the mobilities calculated from the mean square displacement and the velocity autocorrelation functions, respectively, were found to be in good agreement with each other. The calculations demonstrate, for the first time, cation and anion mobilities that fall on separate curves, as functions of ion size, with distinct maxima. This is in complete accord with experimental trends observed in water at 25 °C. The cation mobilities are also in better agreement with the measured values than the calculations done earlier (J. Chem. Phys. 1994, 101, 6964) using the TIP4P model. The mobilities of the halides calculated here for the SPC/E model are however slightly lower than the experimental results. The residence times of water in the hydration shells around an ion are found to decrease dramatically with its size. Stereoscopic pictures show that the structure of the solvent cage around an ion is qualitatively different for the larger ions, implicating both solvent dynamics and structure as important factors in explaining ion mobility in aqueous systems. -

University of Southampton Research Repository Eprints Soton
University of Southampton Research Repository ePrints Soton Copyright © and Moral Rights for this thesis are retained by the author and/or other copyright owners. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the copyright holder/s. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the copyright holders. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given e.g. AUTHOR (year of submission) "Full thesis title", University of Southampton, name of the University School or Department, PhD Thesis, pagination http://eprints.soton.ac.uk UNIVERSITY OF SOUTHAMPTON FACULTY OF NATURAL AND ENVIRONMENTAL SCIENCES Chemistry DEVELOPMENT OF RESONANT INELASTIC X-RAY SCATTERING SPECTROSCOPY FOR 4d AND 5d TRANSITION METAL CATALYSTS Rowena Thomas Thesis for Degree of Doctor of Philosophy APRIL 2013 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF NATURAL AND ENVIRONMENTAL SCIENCES Chemistry Doctor of Philosophy DEVELOPMENT OF RESONANT INELASTIC X-RAY SCATTERING SPECTROSCOPY FOR 4d AND 5d TRANSITION METAL COMPLEXES By Rowena Thomas This research focuses on the development of Resonant Inelastic X-ray Scattering spectroscopy (RIXS) as a tool in homogeneous catalysis for 4d and 5d transition metals. In the RIXS data 2D plots of x-ray emission spectra as a function of absorption were obtained, showing the relationship between the two techniques as well as probing both the unfilled and filled DOS. -

SDS US 2PD Version #: 01 Issue Date: 03-25-2021 1 / 6 Chemical Name Common Name and Synonyms CAS Number % Tungsten Chloride 13283-01-7 100
SAFETY DATA SHEET 1. Identification Product identifier Tungsten Chloride Other means of identification SDS number 2PD Materion Code 2PD CAS number 13283-01-7 Manufacturer/Importer/Supplier/Distributor information Manufacturer Company name Materion Advanced Chemicals Inc. Address 407 N 13th Street 1316 W. St. Paul Avenue Milwaukee, WI 53233 United States Telephone 414.212.0290 E-mail [email protected] Contact person Laura Hamilton Emergency phone number Chemtrec 800.424.9300 2. Hazard(s) identification Physical hazards Not classified. Health hazards Skin corrosion/irritation Category 1 Serious eye damage/eye irritation Category 1 Environmental hazards Not classified. OSHA defined hazards Not classified. Label elements Signal word Danger Hazard statement Causes severe skin burns and eye damage. Causes serious eye damage. Precautionary statement Prevention Do not breathe dust/mist/spray. Wash hands thoroughly after handling. Wear protective gloves/protective clothing/eye protection/face protection. Response IF SWALLOWED: Rinse mouth. Do NOT induce vomiting. IF ON SKIN (or hair): Remove/take off immediately all contaminated clothing. Rinse skin with water/shower. IF INHALED: Remove to fresh air and keep at rest in a position comfortable for breathing. Immediately call a poison center/doctor. Wash/decontaminate removed clothing before reuse. Storage Store locked up. Disposal Dispose of contents/container in accordance with local/regional/national/international regulations. Hazard(s) not otherwise None known. classified (HNOC) Supplemental information None. 3. Composition/information on ingredients Substances Material name: Tungsten Chloride SDS US 2PD Version #: 01 Issue date: 03-25-2021 1 / 6 Chemical name Common name and synonyms CAS number % Tungsten Chloride 13283-01-7 100 4. -

Halides and Halogens. What Do I Need to Know? John Vivari, Nordson EFD
Halides and Halogens. What do I need to know? John Vivari, Nordson EFD Abstract With halogen-containing substances in the public eye due to scrutiny by the European Union and a variety of non- governmental organizations (NGOs) as possible additions to the list of substances banned from electronics, we at EFD have received numerous inquiries from customers asking how this subject will affect them and their processes. Having just overcome the hurdle of RoHS (Restriction of Hazardous Substances), they want to know what halogens and halides are, and what changes they should be prepared for if required to stop using them. Halide-free materials are not new. Some segments of the electronics industry have been sensitive to halides and their significance for decades. This paper will give the reader a working knowledge of halogens and halides. Armed with this education, the reader will be able to make informed decisions when required to use halogen-free materials, either because regulations dictate it or social pressure makes acceptance preferable to resistance. Key Words: halide, halogen, bromine, chlorine, flame retardant, RoHS What are halogens and halides? damage. Brominated flame retardant use is not limited to electronics. It is also in common usage in furniture, At their most basic level, halogens are the electronegative construction materials and textiles. elements in column 17 of the periodic table, including fluorine (F), chlorine, (Cl), bromine (Br), iodine (I) and Other sources of halogens in circuit boards include astatine (At). In electronics fiberglass sizing, epoxy curing agents and accelerators, applications, iodine and resin wetting and de-foaming agents, flux residues, and astatine are rarely if ever contamination from handling. -

SDS EU 2PD Version #: 01 Issue Date: 25-March-2021 1 / 8 Hazard Statements H314 Causes Severe Skin Burns and Eye Damage
SAFETY DATA SHEET SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Name of the substance Tungsten Chloride Identification number 236-293-9 (EC number) Registration number - Document number 2PD Synonyms None. Materion Code 2PD Issue date 25-March-2021 Version number 01 1.3. Details of the supplier of the safety data sheet Supplier Company name Materion Advanced Chemicals Inc. Address 407 N. 13th Street 1316 W. St. Paul Avenue Milwaukee, WI 53233 United States Division Milwaukee Telephone 414.212.0257 e-mail [email protected] Contact person Laura Hamilton 1.4. Emergency telephone number 1.2. Relevant identified uses of the substance or mixture and uses advised against Identified uses Not available. Uses advised against None known. 1.3. Details of the supplier of the safety data sheet Supplier Company name Materion Advanced Chemicals Inc. Address 407 N. 13th Street 1316 W. St. Paul Avenue Milwaukee, WI 53233 United States Division Milwaukee Telephone 414.212.0257 e-mail [email protected] Contact person Laura Hamilton 1.4. Emergency telephone number SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 as amended Hazard summary Causes severe skin burns and eye damage. Causes serious eye damage. 2.2. Label elements Label according to Regulation (EC) No. 1272/2008 as amended Contains: Tungsten Chloride Hazard pictograms Signal word Danger Material name: Tungsten Chloride SDS EU 2PD Version #: 01 Issue date: 25-March-2021 1 / 8 Hazard statements H314 Causes severe skin burns and eye damage. -
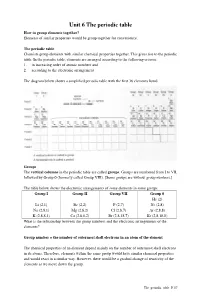
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Safety Data Sheet Material Name: Wcl6 (Tungsten Hexachloride) SDS ID: 320P
Safety Data Sheet Material Name: WCl6 (Tungsten Hexachloride) SDS ID: 320P Section 1 - PRODUCT AND COMPANY IDENTIFICATION Material Name WCl6 (Tungsten Hexachloride) Synonyms TUNGSTEN CHLORIDE; TUNGSTEN HEXACHLORIDE; TUNGSTEN (VI) CHLORIDE Chemical Family metal, halides Product Use semiconductor manufacture Restrictions on Use None known. Details of the supplier of the safety data sheet Entegris, Inc. 129 Concord Road Building 2 Billerica, MA 01821 USA Telephone Number: +1-952-556-4181 Telephone Number: +1-800-394-4083 (toll free within North America) Emergency Telephone Number: CHEMTREC - U.S. - 1-800-424-9300 CHEMTREC - Intl. - 1-703-527-3887 E-mail: [email protected] Section 2 - HAZARDS IDENTIFICATION Classification in accordance with paragraph (d) of 29 CFR 1910.1200. Skin Corrosion/Irritation - Category 1 Serious Eye Damage/Eye Irritation - Category 1 Specific target organ toxicity - Single exposure - Category 3 ( respiratory system ) GHS Label Elements Symbol(s) Signal Word Danger Hazard Statement(s) Causes severe skin burns and eye damage. May cause respiratory irritation. Precautionary Statement(s) Prevention ____________________________________________________________ Page 1 of 9 Issue date: 2020-03-26 Revision 3.4 Print date: 2020-03-26 Safety Data Sheet Material Name: WCl6 (Tungsten Hexachloride) SDS ID: 320P Do not breathe dust. Wash thoroughly after handling. Wear protective gloves/protective clothing/eye protection/face protection. Use only outdoors or in a well-ventilated area. Response Immediately call a POISON CENTER or doctor/physician. IF INHALED: Remove person to fresh air and keep comfortable for breathing. Specific treatment may be needed, see first aid section of Safety Data Sheet. IF ON SKIN (or hair): Take off immediately all contaminated clothing. -

New Route to Synthesize Surface Organometallic Complexes (SOMC): an Approach by Alkylating Halogenated Surface Organometallic Fragments
New Route to Synthesize Surface Organometallic Complexes (SOMC): An Approach by Alkylating Halogenated Surface Organometallic Fragments. Dissertation by Ali Hamieh In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy King Abdullah University of Science and Technology Thuwal, Kingdom of Saudi Arabia February, 2017 2 EXAMINATION COMMITTEE PAGE The dissertation of Ali Hamieh is approved by the examination committee. Committee Chairperson: Professor Jean-Marie Basset Committee Members: Professor Kazuhiro Takanabe, Professor Udo Schwingenschlogl, Professor Joumana Toufaily. 3 © February, 2017 Ali Hamieh All Rights Reserved 4 ABSTRACT New Route to Synthesize Surface Organometallic Complexes (SOMC): An Approach by Alkylating Halogenated Surface Organometallic Fragments. Ali I. Hamieh The aim of this thesis is to explore new simpler and efficient routes for the preparation of surface organometallic complexes (SOMC) for the transformation of small organic molecules to valuable products. The key element in this new route relies on surface alkylation of various halogenated surface coordination complexes or organometallic fragments (SOMF). The first chapter provides an overview on the origin of organometallic compounds, their classical synthesis, characterization and some of their applications In the second chapter, novel silica-supported tungsten oxo-trimethyl complex [(≡Si-O- )W(=O)Me3] was synthesized using the new SOMC synthetic approach. WOCl4 was grafted on the surface of silica, partially dehydroxylated at 700°C (SiO2-700), and [(≡Si-O- )W(=O)Cl3] was produced. The supported complex methylated with ZnMe2 and transformed into [(≡Si-O-)W(=O)Me3], which was fully characterized. It was found that complex [(≡Si-O-)W(=O)Me3] has two conformational isomers at room temperature. -

INORGANIC SYNTHESES Volume 23 Board of Directors
INORGANIC SYNTHESES Volume 23 Board of Directors DUWARD F. SHRIVER Norrhwesrern University HENRY F. HOLZCLAW, JR. University of Nebraska BODIE E. DOUGLAS University of Pirrsburgh JAY H. WORRELL University of Sourh Florida JOHN P. FACKLER, JR. Texas A&M University SMITH L. HOLT, JR. Oklahoma State University Future Volumes 24 JEAN’NE SHREEVE University of Idaho 25 HERBERT D. KAESZ University of California, Los Angeles 26 HARRY R. ALLCOCK Pennsylvania State University 27 STEVEN D. ITTEL E. I. du Ponr de Nemours and Co. 28 ALVIN P. GINSBERG Bell Laboratories 29 ROBERT J. ANGELIC1 Iowa Srare University International Associates MARTIN A. BENNETT Australian National University FAUSTO CALDEWO University of Pisa E. 0. FISCHER Technische Universirar Miinchen SACK LEWIS Cambridge University LAMBERTO MALATESTA University of Milan RENE POILBLANC University of Toulouse HERBERT ROESKY University of Goningen F. G. A. STONE University of Brisrol GEOFFREY WILKINSON Imperial College of Science and Technology AKIO YAMAMOTO Tokyo Kogyo Daigaku (TokyoInstirure of Technology) Editor-in-Chief STANLEY KIRSCHNER Deportment of Chemistry Wayne State Universily Detroit, Michigan INORGANIC SYNTHESES Volume 23 A Wiley-Interscience Publication JOHN WILEY tk SONS New York Chichester Briskne Toronto Singapore Published by John Wiley & Sons, Inc. Copyright 0 1985 by Inorganic Syntheses, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Section 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc. Library of Congress Caralog Number: 39-23015 ISBN 0-471-81873-9 Printed in the United States of America 10 9 8 7 6 5 4 3 2 I HARRYR. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions.