A New Paradigm for Transcription Factor TFIIB Functionality
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

TP53 9606.ENSP00000269305 Tumor Protein P53; Acts As a Tumor
TP53 9606.ENSP00000269305 tumor protein p53; Acts as a tumor suppressor in many tumor types; induces growth arrest or apoptosis depending on the physiological circumstances and cell type. Involved in cell cycle regulation as a trans-activator that acts to negatively regulate cell division by controlling a set of genes required for this process. One of the activated genes is an inhibitor of cyclin-dependent kinases. Apoptosis induction seems to be mediated either by stimulation of BAX and FAS antigen expression, or by repression of Bcl-2 expression. Implicated in Notch signaling cross-over TAF1 9606.ENSP00000276072 TAF1 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 250kDa; Largest component and core scaffold of the TFIID basal transcription factor complex. Contains novel N- and C-terminal Ser/Thr kinase domains which can autophosphorylate or transphosphorylate other transcription factors. Phosphorylates TP53 on 'Thr-55' which leads to MDM2-mediated degradation of TP53. Phosphorylates GTF2A1 and GTF2F1 on Ser residues. Possesses DNA- binding activity. Essential for progression of the G1 phase of the cell cycle SUMO2 9606.ENSP00000405965 SMT3 suppressor of mif two 3 homolog 2 (S. cerevisiae); Ubiquitin-like protein which can be covalently attached to target lysines either as a monomer or as a lysine-linked polymer. Does not seem to be involved in protein degradation and may function as an antagonist of ubiquitin in the degradation process. Plays a role in a number of cellular processes such as nuclear transport, DNA -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

Zinc Finger Protein 185 Is a Liver Metastasis‑Associated Factor in Colon Cancer Patients
MOLECULAR AND CLINICAL ONCOLOGY 2: 709-713 Zinc finger protein 185 is a liver metastasis‑associated factor in colon cancer patients DAISUKE FURUKAWA1, TSUYOSHI CHIJIWA2, MASAHIRO MATSUYAMA1, MASAYA MUKAI3, EI-ICHI MATSUO4, OSAMU NISHIMURA5, KENJI KAWAI6, HIROSHI SUEMIZU2, NOBUYOSHI HIRAOKA7, TOSHIO NAKAGOHRI1, SEIEI YASUDA1 and MASATO NAKAMURA8 1Department of Surgery, Tokai University School of Medicine, Isehara, Kanagawa 259-1193; 2Laboratory Animal Research Department, Central Institute for Experimental Animals, Kawasaki, Kanagawa 210-0821; 3Department of Surgery, Tokai University Hachioji Hospital, Hachioji, Tokyo 192-0032; 4Global Application Development Center, Analytical and Measuring Instruments Division, Shimadzu Corporation, Kyoto 604-8511; 5The Integrated Center for Mass Spectrometry, Graduate School of Medicine, Kobe University, Kobe, Hyogo 650-0017; 6Pathological Analysis Center, Central Institute for Experimental Animals, Kawasaki, Kanagawa 210-0821; 7Division of Molecular Pathology, National Cancer Center Research Institute, Tokyo 104-0045; 8 Department of Regenerative Medicine, Tokai University School of Medicine, Isehara, Kanagawa 259-1193, Japan Received February 20, 2014; Accepted March 25, 2014 DOI: 10.3892/mco.2014.298 Abstract. LIM domain proteins are involved in several respectively). Therefore, ZNF185 expression was found to be fundamental biological processes, including cell lineage an independent indicator of liver metastasis and prognosis in specification, cytoskeleton organization and organ devel- patients with colon cancer. opment. Zinc finger protein 185 (ZNF185) is one of the LIM domain proteins considered to be involved in the regula- Introduction tion of cellular differentiation and/or proliferation. However, the detailed functions and properties of ZNF185 in the multi- Colorectal cancer is among the three leading causes of step process of cancer biology have not yet been elucidated. -

Tubular P53 Regulates Multiple Genes to Mediate AKI
BASIC RESEARCH www.jasn.org Tubular p53 Regulates Multiple Genes to Mediate AKI † † † † † Dongshan Zhang,* Yu Liu,* Qingqing Wei, Yuqing Huo, Kebin Liu, Fuyou Liu,* and † Zheng Dong* *Departments of Emergency Medicine and Nephrology, Second Xiangya Hospital, Central South University, Changsha, Hunan, China; and †Department of Cellular Biology and Anatomy, Vascular Biology Center and Department of Biochemistry and Molecular Biology, Georgia Regents University and Charlie Norwood Veterans Affairs Medical Center, Augusta, Georgia ABSTRACT A pathogenic role of p53 in AKI was suggested a decade ago but remains controversial. Indeed, recent work indicates that inhibition of p53 protects against ischemic AKI in rats but exacerbates AKI in mice. One intriguing possibility is that p53 has cell type-specific roles in AKI. To determine the role of tubular p53, we generated two conditional gene knockout mouse models, in which p53 is specifically ablated from proximal tubules or other tubular segments, including distal tubules, loops of Henle, and medullary collecting ducts. Proximal tubule p53 knockout (PT-p53-KO) mice were resistant to ischemic and cisplatin nephrotoxic AKI, which was indicated by the analysis of renal function, histology, apoptosis, and inflammation. However, other tubular p53 knockout (OT-p53-KO) mice were sensitive to AKI. Mechanis- tically, AKI associated with the upregulation of several known p53 target genes, including Bax, p53- upregulated modulator of apoptosis-a, p21, and Siva, and this association was attenuated in PT-p53-KO mice. In global expression analysis, ischemic AKI induced 371 genes in wild-type kidney cortical tissues, but the induction of 31 of these genes was abrogated in PT-p53-KO tissues. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

PDF-Document
Supplementary Material Investigating the role of microRNA and Transcription Factor co-regulatory networks in Multiple Sclerosis pathogenesis Nicoletta Nuzziello1, Laura Vilardo2, Paride Pelucchi2, Arianna Consiglio1, Sabino Liuni1, Maria Trojano3 and Maria Liguori1* 1National Research Council, Institute of Biomedical Technologies, Bari Unit, Bari, Italy 2National Research Council, Institute of Biomedical Technologies, Segrate Unit, Milan, Italy 3Department of Basic Sciences, Neurosciences and Sense Organs, University of Bari, Bari, Italy Supplementary Figure S1 Frequencies of GO terms and canonical pathways. (a) Histogram illustrates the GO terms associated to assembled sub-networks. (b) Histogram illustrates the canonical pathways associated to assembled sub-network. a b Legends for Supplementary Tables Supplementary Table S1 List of feedback (FBL) and feed-forward (FFL) loops in miRNA-TF co-regulatory network. Supplementary Table S2 List of significantly (adj p-value < 0.05) GO-term involved in MS. The first column (from the left) listed the GO-term (biological processes) involved in MS. For each functional class, the main attributes (gene count, p-value, adjusted p-value of the enriched terms for multiple testing using the Benjamini correction) have been detailed. In the last column (on the right), we summarized the target genes involved in each enriched GO-term. Supplementary Table S3 List of significantly (adj p-value < 0.05) enriched pathway involved in MS. The first column (from the left) listed the enriched pathway involved in MS. For each pathway, the main attributes (gene count, p-value, adjusted p-value of the enriched terms for multiple testing using the Benjamini correction) have been detailed. In the last column (on the right), we summarized the target genes involved in each enriched pathway. -

Inflammatory and Adipogenic Stromal Subpopulations in Visceral Adipose Tissue Of
RESEARCH COMMUNICATION Identification of functionally distinct fibro- inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice Chelsea Hepler1†, Bo Shan1, Qianbin Zhang1, Gervaise H Henry2, Mengle Shao1, Lavanya Vishvanath1, Alexandra L Ghaben1, Angela B Mobley3, Douglas Strand2, Gary C Hon4, Rana K Gupta1* 1Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, United States; 2Department of Urology, University of Texas Southwestern Medical Center, Dallas, United States; 3Department of Immunology, University of Texas Southwestern Medical Center, Dallas, United States; 4Cecil H. and Ida Green Center for Reproductive Biology Sciences, Division of Basic Reproductive Biology Research, Department of Obstetrics and Gynecology, University of Texas Southwestern Medical Center, Dallas, United States Abstract White adipose tissue (WAT) remodeling is dictated by coordinated interactions between adipocytes and resident stromal-vascular cells; however, the functional heterogeneity of adipose stromal cells has remained unresolved. We combined single-cell RNA-sequencing and FACS to identify and isolate functionally distinct subpopulations of PDGFRb+ stromal cells within visceral WAT of adult mice. LY6C- CD9- PDGFRb+ cells represent highly adipogenic visceral adipocyte precursor cells (‘APCs’), whereas LY6C+ PDGFRb+ cells represent fibro-inflammatory progenitors (‘FIPs’). FIPs lack adipogenic capacity, display pro-fibrogenic/pro-inflammatory *For correspondence: -
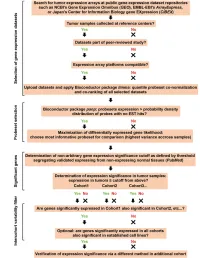
Supplementary Data
SUPPLEMENTAL INFORMATION A study restricted to chemokine receptors as well as a genome-wide transcript analysis uncovered CXCR4 as preferentially expressed in Ewing's sarcoma (Ewing's sarcoma) cells of metastatic origin (Figure 4). Transcriptome analyses showed that in addition to CXCR4, genes known to support cell motility and invasion topped the list of genes preferentially expressed in metastasis-derived cells (Figure 4D). These included kynurenine 3-monooxygenase (KMO), galectin-1 (LGALS1), gastrin-releasing peptide (GRP), procollagen C-endopeptidase enhancer (PCOLCE), and ephrin receptor B (EPHB3). KMO, a key enzyme of tryptophan catabolism, has not been linked to metastasis. Tryptophan and its catabolites, however, are involved in immune evasion by tumors, a process that can assist in tumor progression and metastasis (1). LGALS1, GRP, PCOLCE and EPHB3 have been linked to tumor progression and metastasis of several cancers (2-4). Top genes preferentially expressed in L-EDCL included genes that suppress cell motility and/or potentiate cell adhesion such as plakophilin 1 (PKP1), neuropeptide Y (NPY), or the metastasis suppressor TXNIP (5-7) (Figure 4D). Overall, L-EDCL were enriched in gene sets geared at optimizing nutrient transport and usage (Figure 4D; Supplementary Table 3), a state that may support the early stages of tumor growth. Once tumor growth outpaces nutrient and oxygen supplies, gene expression programs are usually switched to hypoxic response and neoangiogenesis, which ultimately lead to tumor egress and metastasis. Accordingly, gene sets involved in extracellular matrix remodeling, MAPK signaling, and response to hypoxia were up-regulated in M-EDCL (Figure 4D; Supplementary Table 4), consistent with their association to metastasis in other cancers (8, 9). -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility.