Biologics CDMO Trends and Opportunities in China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

China Multi Asset Income Fund - 3-Year Class USD Fund Volatility
Principal China Multi Asset Income Fund - 3-year Class USD Fund Volatility 31 August 2021 10.83Low Lipper Analytics 15 Aug 2021 Fund Objective Fund Performance The Fund aims to provide income and 60% moderate capital growth through investments in one CIS, which invests 50% primarily in a diversified portfolio of securities related to China. 40% Currency: ISIN Code: 30% USD MYU1002GD004 20% Bloomberg Ticker: CIMCMUS MK 10% 0% Fund Information 7 7 8 8 8 8 8 8 9 9 9 9 9 9 0 0 0 0 0 0 1 1 1 1 1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 -2 -2 -2 -2 -2 -2 -2 -2 -2 -2 t c b r n g t c b r n l p v n r y l p v n b r n g c e e p u u c e e p u Ju e o a a a Ju e o a e p u u - 10% O D F A J A O D F A J S N J M M S N J F A J A Domicile Malaysia - 20% Base Currency US Dollar (USD) Fund Inception 3 July 2017 Fund Benchmark Benchmark The Fund adheres to the Past performance does not guarantee future results. Asset allocation and diversification do not ensure a profit or protect against a loss. benchmark of the Target Fund for performance Cumulative Performance (%) comparison. The Since YTD 1 Month 3 Months 6 Months 1-Year 3-Year 5-Year Inception benchmark of the Target Fund -8.70 -0.21 -9.33 -10.95 -1.42 17.94 N/A 19.79 Fund is 50% MSCI China Net + 50% Markit Benchmark/Target Return -4.47 0.22 -7.23 -7.95 2.29 22.31 N/A 33.59 iBoxx Asia Local Bond Index China Offshore for Calendar Year Returns (%) 2020 2019 2018 2017 2016 2015 comparison purpose. -

Crystal Reports
South Orlando Baptist Church LIBRARY 1.7 BIBLIOGRAPHY WITH SUMMARY Page 1 Friday, August 15, 2014 Search Criteria: Classification starting with 'JF' Classification Author Media Type Call Title Accession # Summary Copyright Subject 1 Subject 2 JF Adler, Susan S. Book Adl Meet Samantha : an American girl 6375 In 1904, nine-year-old Samantha, an orphan living with her wealthy grandmother, and her servant friend Nellie have a midnight adventure when they try to find out what has happened 1998 C. Friendship -- Fiction Orphans -- Fiction JF Adler, Susan S. Book Adl Samantha learns a lesson : a school story 6376 When nine-year-old Nellie begins to attend school, Samantha determines to help her with her schoolwork and learns a great deal herself about what it is like to be a poor child and 1998 C. Schools -- Fiction Friendship -- Fiction JF TOO TALL Adshead, Paul Book Ads Puzzle island 7764 The reader is asked to find hidden animals in the pictures and identify a mysterious creature believed to be extinct. 1995 C. Picture puzzles Animals -- Fiction JF Aesop Book Aes Aesop's fables (Great Illustrated Classics) 6769 An illustrated adaptation of fables first told by the Greek slave Aesop. 2005 C. Aesop's fables -- Adaptations Fables JF Alvarez, Julia Book Alv How Tía Lola came to visit stay 8311 Although ten-year-old Miguel is at first embarrassed by his colorful aunt, Tia Lola, when she comes to Vermont from the Dominican Republic to stay with his mother, his sister, and 2001 C. Aunts -- Fiction Dominican Americans -- Fiction JF Amato, Mary Book Ama The word eater 6381 Lerner Chanse, a new student at Cleveland Park Middle School, finds a worm that magically makes things disappear, and she hopes it will help her fit in, or get revenge, at her hate 2000 C. -

Hang Seng Indexes Announces Index Review Results
14 August 2020 Hang Seng Indexes Announces Index Review Results Hang Seng Indexes Company Limited (“Hang Seng Indexes”) today announced the results of its review of the Hang Seng Family of Indexes for the quarter ended 30 June 2020. All changes will take effect on 7 September 2020 (Monday). 1. Hang Seng Index The following constituent changes will be made to the Hang Seng Index. The total number of constituents remains unchanged at 50. Inclusion: Code Company 1810 Xiaomi Corporation - W 2269 WuXi Biologics (Cayman) Inc. 9988 Alibaba Group Holding Ltd. - SW Removal: Code Company 83 Sino Land Co. Ltd. 151 Want Want China Holdings Ltd. 1088 China Shenhua Energy Co. Ltd. - H Shares The list of constituents is provided in Appendix 1. The Hang Seng Index Advisory Committee today reviewed the fast expanding innovation and new economy sectors in the Hong Kong capital market and agreed with the proposal from Hang Seng Indexes to conduct a comprehensive study on the composition of the Hang Seng Index. This holistic review will encompass various aspects including, but not limited to, composition and selection of constituents, number of constituents, weightings, and industry and geographical representation, etc. The underlying aim of the study is to ensure the Hang Seng Index continues to serve as the most representative and important benchmark of the Hong Kong stock market. Hang Seng Indexes will report its findings and propose recommendations to the Advisory Committee within six months. The number of constituents of the Hang Seng Index may increase during this period. Hang Seng Indexes Announces Index Review Results /2 2. -

Greater China 2019
IR Magazine Awards – Greater China 2019 Winners and nominees AWARDS BY RESEARCH Best overall investor relations (large cap) ANTA Sports Products China Resources Beer WINNER China Telecom China Unicom Shenzhou International Group Holdings Best overall investor relations (small to mid-cap) Alibaba Pictures Group Far East Consortium International WINNER Health and Happiness H&H International Holdings Li-Ning NetDragon Websoft Holdings Best investor relations officer (large cap) ANTA Sports Products Suki Wong Cathay Financial Holdings Yajou Chang & Sophia Cheng China Resources Beer Vincent Tse WINNER China Telecom Lisa Lai China Unicom Jacky Yung Best investor relations officer (small to mid-cap) Agile Group Holdings Samson Chan BizLink Holding Tom Huang Far East Consortium International Venus Zhao WINNER Li-Ning Rebecca Zhang Yue Yuen Industrial (Holdings) Olivia Wang Best IR by a senior management team Maggie Wu, CFO & Daniel Zhang, Alibaba Group CEO Tomakin Lai Po-sing, CFO & China Resources Beer Xiaohai Hou, CEO Xiaochu Wang, CEO & Zhu WINNER China Unicom Kebing, CFO Wai Hung Boswell Cheung, CFO & Far East Consortium International David Chiu, Chairman & CEO Ma Jianrong, CEO & Cun Bo Wang, Shenzhou International Group Holdings CFO AWARDS BY REGION Best in region: China Alibaba Pictures Group ANTA Sports Products China Resources Beer WINNER China Telecom China Unicom Shenzhou International Group Holdings Best in region: Hong Kong AIA Group Far East Consortium International WINNER Health and Happiness H&H International Holdings Yue Yuen -

HSBC Emerging Market Sustainable Equity UCITS ETF
Sustainably Invested MX-PE HSBC ETFs PLC - HSBC Emerging Market Sustainable Equity UCITS ETF HSEF LN Aug 31 2021 Fund IE00BKY59G90 Aug 31 2021 Fund Objective and Strategy Investment Objective The Fund aims to track as closely as possible the returns of the FTSE Emerging ESG Low Carbon Select Index (the “Index”). The Fund will invest in or gain exposure to shares of companies which make up the Index. Investment Policy The Index seeks to achieve a reduction in carbon emissions and fossil fuel reserves exposure, improvement of the FTSE Russell ESG rating and also applies the United Nations Global Compact exclusionary criteria.The Fund will be passively managed and will aim to invest in the shares of the companies in generally the same proportion as in the Index. However, there may be circumstances when it is not possible or practical for the Fund to invest in all constituents of the Index. The Fund can gain exposure by using other investments such as depositary receipts, derivatives or funds.The Fund may invest up to 35% in securities from a single issuer during exceptional market conditions.The Fund may invest up to 10% in funds and up to 10% in total return swaps and contracts for difference.See the Prospectus for a full description of the investment objectives and derivative usage. Since Inception Performance (%) Fund Details UCITS V Compliant Yes 130 Fund Reference Benchmark 125 Distribution Type Accumulating 120 2 115 Ongoing Charge Figure 0.180% 110 105 Share Class Base Currency USD 100 95 90 Domicile Ireland ISIN IE00BKY59G90 Share -

HSBC Global Investment Funds CHINESE EQUITY Monthly Report 31 August 2021 | Share Class IC
HSBC Global Investment Funds CHINESE EQUITY Monthly report 31 August 2021 | Share class IC Share Class Details Investment objective Key metrics The Fund aims to provide long term capital growth by investing in a portfolio of Chinese shares. NAV per Share USD 163.81 Performance 1 month -0.91% Investment strategy Volatility 3 years 21.14% Fund facts In normal market conditions, the Fund will invest at least 90% of its assets in shares (or securities UCITS V compliant Yes similar to shares) of companies of any size that are based in, or carry out the larger part of their business activities in, China, including Hong Kong SAR. The Fund can invest up to 70% in China Dividend treatment Accumulating A and China B-shares. For China A-shares, up to 70% through the Shanghai-Hong Kong Stock Dealing frequency Daily Connect and/or the Shenzhen-Hong Kong Stock Connect, and up to 50% in CAAPs. The Fund Valuation Time 17:00 Luxembourg may invest up to 10% of its assets in other funds, including HSBC funds. The Fund will not invest more than 10% of its assets in Real Estate Investment Trusts. See the Prospectus for a full Share Class Base Currency USD description of the investment objectives and derivative usage. Domicile Luxembourg Inception date 27 July 2005 Main risks Fund Size USD 1,139,298,382 Reference 100% MSCI China 10/40 • The Fund's unit value can go up as well as down, and any capital invested in the Fund may benchmark Net be at risk. • The value of investible securities can change over time due to a wide variety of factors, Managers Caroline Yu Maurer including but not limited to: political and economic news, government policy, changes in Fees and expenses demographics, cultures and populations, natural or human-caused disasters etc. -

South Orlando Baptist Church LIBRARY RECORDS by SUBJECT
South Orlando Baptist Church LIBRARY RECORDS BY SUBJECT Page 1 Friday, November 15, 2013 Find all records where any portion of Basic Fields (Title, Author, Subjects, Summary, & Comments) like '' Subject Title Classification Author Accession # Abandoned children Fiction A cry in the dark (Summerhill Secrets #5) JF Lewis, Beverly 6543 Abandoned houses Fiction Julia's hope (The Wortham Family Series #1) F Kelly, Leisha 5343 Abolitionists Frederick Douglass (Black Americans of Achievement) JB Russell, Sharman Apt. 8173 Abortion Fiction Choice summer (Nikki Sheridan Series #1) YA F Brinkerhoff, Shirley 7970 Shades of blue F Kingsbury, Karen 8823 Tilly : the novel F Peretti, Frank E. 6204 Abortion Moral and ethical aspects Abortion : a rational look at an emotional issue 241 Sproul, R. C. (Robert Charles) 1296 Abraham (Biblical patriarch) Abraham and his big family [videorecording] VHS C220.95 Walton, John H. 5221 Abraham, man of faith J221.92 Rives, Elsie 1513 Created to be God's friend : how God shapes those He loves 248.4 Blackaby, Henry T. 4008 Dragons (Face to face) J398.24 Dixon, Dougal 8517 The story of Abraham (Great Bible Stories) C221.92 Nodel, Maxine 3724 Abused children Fiction Looking for Cassandra Jane F Carlson, Melody 6052 Abused wives Fiction A place called Wiregrass F Morris, Michael 7881 Wings of a dove F Bush, Beverly 2498 Abused women Fiction Sharon's hope F Coyle, Neva 3706 Acadians Fiction The beloved land (Song of Acadia #5) F Oke, Janette 3910 The distant beacon (Song of Acadia #4) F Oke, Janette 3690 The innocent libertine (Heirs of Acadia #2) F Bunn, T. -

Materials and Structures Toward Soft Electronics
REVIEW Soft Electronics www.advmat.de Materials and Structures toward Soft Electronics Chunfeng Wang, Chonghe Wang, Zhenlong Huang, and Sheng Xu* very well,[10–14] and therefore this article Soft electronics are intensively studied as the integration of electronics with will mainly focus on the stretchable aspect dynamic nonplanar surfaces has become necessary. Here, a discussion of the of soft electronic devices. strategies in materials innovation and structural design to build soft elec- The implications of soft electronics inte- tronic devices and systems is provided. For each strategy, the presentation grating with nonplanar objects are multi- focuses on the fundamental materials science and mechanics, and example fold. First, the intimate contact between the device and the nonplanar object will device applications are highlighted where possible. Finally, perspectives on allow high-quality data to be collected.[15] the key challenges and future directions of this field are presented. With rigid electronics, air gaps at the interface between the device and the object reduce the contact area, and can 1. Introduction potentially introduce noise and artifacts, which compromise signal quality.[5] Second, foldable, low-profile devices can enable Nonplanar surfaces, either static (e.g., complex shaped objects) mobile and distributed sensing, which hold great promise for or dynamic (e.g., biology), are prevalent in nature. Soft elec- Internet-of-Things technology.[16] Finally, in the area of medical tronics that allow interfacing with nonplanar -
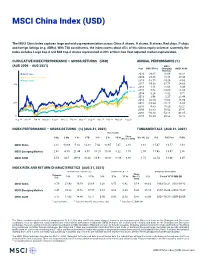
MSCI China Index (USD)
MSCI China Index (USD) The MSCI China Index captures large and mid cap representation across China A shares, H shares, B shares, Red chips, P chips and foreign listings (e.g. ADRs). With 730 constituents, the index covers about 85% of this China equity universe. Currently, the index includes Large Cap A and Mid Cap A shares represented at 20% of their free float adjusted market capitalization. CUMULATIVE INDEX PERFORMANCE — GROSS RETURNS (USD) ANNUAL PERFORMANCE (%) (AUG 2006 – AUG 2021) MSCI Year MSCI China Emerging MSCI ACWI Markets MSCI China 2020 29.67 18.69 16.82 MSCI Emerging Markets 2019 23.66 18.88 27.30 MSCI ACWI 2018 -18.75 -14.24 -8.93 400 2017 54.33 37.75 24.62 366.92 2016 1.11 11.60 8.48 324.55 2015 -7.62 -14.60 -1.84 2014 8.26 -1.82 4.71 2013 3.96 -2.27 23.44 252.73 2012 23.10 18.63 16.80 200 2011 -18.24 -18.17 -6.86 2010 4.83 19.20 13.21 2009 62.63 79.02 35.41 2008 -50.83 -53.18 -41.85 50 2007 66.24 39.82 12.18 Aug 06 Nov 07 Feb 09 May 10 Aug 11 Nov 12 Feb 14 May 15 Aug 16 Nov 17 Feb 19 May 20 Aug 21 INDEX PERFORMANCE — GROSS RETURNS (%) (AUG 31, 2021) FUNDAMENTALS (AUG 31, 2021) ANNUALIZED Since 1 Mo 3 Mo 1 Yr YTD 3 Yr 5 Yr 10 Yr Dec 31, 1992 Div Yld (%) P/E P/E Fwd P/BV MSCI China 0.01 -13.69 -5.00 -12.18 7.42 10.97 7.47 2.18 1.61 15.47 13.57 1.91 MSCI Emerging Markets 2.65 -4.00 21.49 3.07 10.25 10.80 5.22 7.75 2.07 15.98 13.07 2.00 MSCI ACWI 2.53 4.67 29.18 16.24 14.91 14.88 11.86 8.88 1.71 22.54 18.46 3.07 INDEX RISK AND RETURN CHARACTERISTICS (AUG 31, 2021) ANNUALIZED STD DEV (%) 2 SHARPE RATIO 2 , 3 MAXIMUM DRAWDOWN Turnover Since 1 3 Yr 5 Yr 10 Yr 3 Yr 5 Yr 10 Yr Dec 31, (%) Period YYYY-MM-DD (%) 1992 MSCI China 8.79 21.43 19.19 20.89 0.38 0.57 0.42 0.14 88.63 1993-12-31—2001-09-12 MSCI Emerging Markets 8.45 19.23 16.52 17.57 0.54 0.63 0.34 0.33 65.14 2007-10-29—2008-10-27 MSCI ACWI 3.17 17.93 14.46 13.77 0.80 0.95 0.83 0.47 58.06 2007-10-31—2009-03-09 1 Last 12 months 2 Based on monthly gross returns data 3 Based on ICE LIBOR 1M The China mainland equity market is comprised of A, B, H, Red chip and P chip share classes. -

Swedish Delight IKEA Opens Its Doors in Penang Story by Edmund Lee Modern and Intelligent Township
槟主办 2020世界 科技大会 《珍珠快讯》 封面版 April 1 – 15, 2019 Swedish delight IKEA opens its doors in Penang Story by Edmund Lee modern and intelligent township. Pix by Darwina Mohd Daud “Batu Kawan, which was for- merly underdeveloped, is now FROM affordable and quality growing into an exciting town- home furnishings to the delicious ship, with 4,017 residential and meatballs. commercial units besides having All these are now available for facilities such as IKEA, KDU Penangites and people in the University College and Design northern region when Swedish Village. furniture giant IKEA opened its “Leading property developers store in Batu Kawan on March such as EcoWorld, Paramount, 14. Aspen Group and Penang Devel- Previously, IKEA customers in opment Corporation (PDC) par- the northern region had to travel ticipated in the development of to Selangor to shop for IKEA’s Batu Kawan,” Chow said in his affordable furniture and home speech during the opening cere- furnishings. mony. Now with the official opening Also present were Finance of the fourth IKEA store in the Minister Lim Guan Eng, IKEA country here, it brings all those Southeast Asia managing director items the firm is well-known for almost right to our doorstep. Chow and Lim with Pathmalingam Three other IKEA stores are (third from left) and Roejkjaer located in Cheras and Damansara (fourth from left) during the grand opening of IKEA Batu in Selangor and Tebrau, Johor. Kawan. The latest IKEA store has 470,146sq ft of retail space. Christian Roejkjaer and IKEA Besides its home furnishings, Batu Kawan store manager A. IKEA’s food outlets are well Pathmalingam. -

Chinese Equity
Chinese Equity Monthly Update April 2021 Performance (% Total Return) Periods ended April 30, 20211 1 Month YTD Since Inception2 HL Chinese Equity (Gross) 3.39 2.70 2.70 HL Chinese Equity (Net) 3.31 2.37 2.37 MSCI China All Shares Index3,4 2.32 0.79 0.79 Portfolio Positioning (% Weight) Investment Perspectives MSCI Market China All As COVID-19 remains well controlled within China and travel Sector HL CE Shares (Under) / Over restrictions are eased, domestic travel seems poised to rebound Industrials 18.5 7.5 during the longest holiday in the first half of this year, the five-day period beginning on May 1 (known as Labor Day). This augurs well Health Care 14.9 8.9 for leisure- and travel-related businesses. Cons Discretionary 29.9 25.1 Portfolio Cash 1.7 – For US investors in Chinese securities, the Holding Foreign Companies Accountable Act imposed in the waning days of the Trump Info Technology 9.3 8.3 administration is a slow-moving threat—but one from which we have nonetheless chosen to insulate the portfolio altogether. To side-step Comm Services 14.5 14.1 the risk (which we still judge to be low) that ADRs would be de-listed from US exchanges in 2023 either because of non-compliant auditing Utilities 1.5 2.0 standards or purported connections to the Chinese military, we exchanged the four such ADR holdings for the underlying ordinary Energy 0.0 1.3 shares, following their secondary listing in Hong Kong. The strategy no longer holds any US-listed ADRs. -

Ireland Asia Report Ireland Asia Report Table of Contents
Ireland Asia Report Ireland Asia Report Table of Contents Ireland as an FDI location for Asian companies Martin Shanahan, CEO, IDA Ireland 2 Ireland’s open economy and its role in the Future of Global Trade Alan Duffy, CEO & Head of Banking, HSBC Ireland 4 An ideal gateway to Europe William Wu, Managing Director, Head of Corporate Banking Yangtze Delta and Western China at HSBC China 7 Asia Market Profiles 8 Mainland China and WuXi Biologics 8 Hong Kong and Goshawk 10 Japan and Indeed.com 12 India and NIIT 14 South Korea and SK Biotek 16 ASEAN and Keppel 18 The Ireland advantage for funds 20 Ireland – a well-rounded destination for people and investment, Chen Tian, Head of China Desk, Continental Europe, Commercial Banking, HSBC 22 Page 1 Ireland Asia Report Ireland Asia Report Ireland as an FDI location for Asian Companies Yet although there may be some unpredictable times Martin Shanahan, ahead, I remain confident in Ireland’s competitiveness CEO IDA Ireland and attractiveness as a place to do business. From regular recent contact between IDA Ireland and many of the leading multinationals here, I know how resilient these companies’ Irish operations are. I believe the supports that Ireland, as a nation, puts in place to welcome international investment are a key factor in this durability. The disruption to supply chains brought about by the We are all still coming to terms with crisis has highlighted how important it is to manage risk and avoid overdependency on a single source. If the shock that the Covid-19 crisis we apply this to an Irish context, it means not relying has delivered to our economy and on any one geographical region or industry sector for society.