Genotypes and Phenotypes of a Family with a Deaf Child Carrying Combined Heterozygous Mutations in SLC26A4 and GJB3 Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Rare Missense Mutation in GJB3 (Cx31g45e) Is Associated with a Unique Cellular Phenotype Resulting in Necrotic Cell Death Easton, J
University of Dundee A rare missense mutation in GJB3 (Cx31G45E) is associated with a unique cellular phenotype resulting in necrotic cell death Easton, J. A.; Alboulshi, A. K.; Kamps, M. A. F.; Brouns, G. H.; Broers, M. R.; Coull, B. J.; Oji, V.; van Geel, M.; van Steensel, M. A. M.; Martin, P. E. Published in: Experimental Dermatology DOI: 10.1111/exd.13542 Publication date: 2018 Document Version Peer reviewed version Link to publication in Discovery Research Portal Citation for published version (APA): Easton, J. A., Alboulshi, A. K., Kamps, M. A. F., Brouns, G. H., Broers, M. R., Coull, B. J., ... Martin, P. E. (2018). A rare missense mutation in GJB3 (Cx31G45E) is associated with a unique cellular phenotype resulting in necrotic cell death. Experimental Dermatology. https://doi.org/10.1111/exd.13542 General rights Copyright and moral rights for the publications made accessible in Discovery Research Portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from Discovery Research Portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. -

Nrf2 Modulates Host Defense During Streptococcus Pneumoniae Pneumonia in Mice
Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice This information is current as John C. Gomez, Hong Dang, Jessica R. Martin and Claire of September 28, 2021. M. Doerschuk J Immunol published online 26 August 2016 http://www.jimmunol.org/content/early/2016/08/26/jimmun ol.1600043 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/08/26/jimmunol.160004 Material 3.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published August 26, 2016, doi:10.4049/jimmunol.1600043 The Journal of Immunology Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice John C. Gomez,*,† Hong Dang,†,‡ Jessica R. Martin,*,† and Claire M. Doerschuk*,†,x Nrf2 regulates the transcriptional response to oxidative stress. These studies tested the role of Nrf2 during Streptococcus pneumoniae pneumonia and identified Nrf2-dependent genes and pathways in lung tissue and in recruited neutrophils. -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Reproductionresearch
REPRODUCTIONRESEARCH Gap junctions are essential for murine primordial follicle assembly immediately before birth Zhen Teng*, Chao Wang*, Yijing Wang, Kun Huang, Xi Xiang, Wanbao Niu, Lizhao Feng, Lihua Zhao, Hao Yan and Hua Zhang State Key Laboratory of Agro-Biotechnology, College of Biological Science, China Agricultural University, Beijing 100193, China Correspondence should be addressed to C Wang; Email: [email protected] *(Z Teng and C Wang contributed equally to this work) Abstract The reserve of primordial follicles determines the reproductive ability of the female mammal over its reproductive life. The primordial follicle is composed of two types of cells: oocytes and surrounding pre-granulosa cells. However, the underlying mechanism regulating primordial follicle assembly is largely undefined. In this study, we found that gap junction communication (GJC) established between the ovarian cells in the perinatal mouse ovary may be involved in the process. First, gap junction structures between the oocyte and surrounding pre-granulosa cells appear at about 19.0 dpc (days post coitum). As many as 12 gap junction-related genes are upregulated at birth, implying that a complex communication may exist between ovarian cells, because specifically silencing the genes of individual gap junction proteins, such as Gja1, Gja4 or both, has no influence on primordial follicle assembly. On the other hand, non-specific blockers of GJC, such as carbenoxolone (CBX) and 18a-glycyrrhetinic acid (AGA), significantly inhibit mouse primordial follicle assembly. We proved that the temporal window for establishment of GJC in the fetal ovary is from 19.5 dpc to 1 dpp (days postpartum). In addition, the expression of ovarian somatic cell (OSC)-specific genes, such as Notch2, Foxl2 and Irx3, was negatively affected by GJC blockers, whereas oocyte-related genes, such as Ybx2, Nobox and Sohlh1, were hardly affected, implying that the establishment of GJC during this period may be more important to OSCs than to oocytes. -

Altered Physiological Functions and Ion Currents in Atrial Fibroblasts From
Physiological Reports ISSN 2051-817X ORIGINAL RESEARCH Altered physiological functions and ion currents in atrial fibroblasts from patients with chronic atrial fibrillation Claire Poulet1, Stephan Kunzel€ 1, Edgar Buttner€ 1, Diana Lindner2, Dirk Westermann2 & Ursula Ravens1 1 Department of Pharmacology and Toxicology, Medical Faculty Carl-Gustav-Carus, TU Dresden, Dresden, Germany 2 Department of General and Interventional Cardiology, University Heart Center Hamburg Eppendorf, Hamburg, Germany Keywords Abstract Atrial fibrillation, electrophysiology, fibroblasts. The contribution of human atrial fibroblasts to cardiac physiology and patho- physiology is poorly understood. Fibroblasts may contribute to arrhythmogen- Correspondence esis through fibrosis, or by directly altering electrical activity in Claire Poulet, Imperial College London, Imperial cardiomyocytes. The objective of our study was to uncover phenotypic differ- Centre for Translational and Experimental ences between cells from patients in sinus rhythm (SR) and chronic atrial fib- Medicine, Hammersmith Campus, Du Cane rillation (AF), with special emphasis on electrophysiological properties. We Road, London W12 0NN, UK isolated fibroblasts from human right atrial tissue for patch-clamp experi- Tel: +44 207 594 2738 Fax: +44 207 594 3653 ments, proliferation, migration, and differentiation assays, and gene expression E-mail: [email protected] profiling. In culture, proliferation and migration of AF fibroblasts were strongly impaired but differentiation into myofibroblasts was increased. This Present Addresses was associated with a higher number of AF fibroblasts expressing functional Claire Poulet, Imperial College London, Nav1.5 channels. Strikingly Na+ currents were considerably larger in AF cells. National Heart and Lung Institute, London, UK Blocking Na+ channels in culture with tetrodotoxin did not affect prolifera- tion, migration, or differentiation in neither SR nor AF cells. -

Abstracts from the 51St European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 27:870–1041 https://doi.org/10.1038/s41431-019-0408-3 MEETING ABSTRACTS Abstracts from the 51st European Society of Human Genetics Conference: Electronic Posters © European Society of Human Genetics 2019 June 16–19, 2018, Fiera Milano Congressi, Milan Italy Sponsorship: Publication of this supplement was sponsored by the European Society of Human Genetics. All content was reviewed and approved by the ESHG Scientific Programme Committee, which held full responsibility for the abstract selections. Disclosure Information: In order to help readers form their own judgments of potential bias in published abstracts, authors are asked to declare any competing financial interests. Contributions of up to EUR 10 000.- (Ten thousand Euros, or equivalent value in kind) per year per company are considered "Modest". Contributions above EUR 10 000.- per year are considered "Significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal Genetics then compared this data to de novo cases where research based PO studies were completed (N=57) in NY. E-P01.01 Results: MFSIQ (66.4) for familial deletions was Parent of origin in familial 22q11.2 deletions impacts full statistically lower (p = .01) than for de novo deletions scale intelligence quotient scores (N=399, MFSIQ=76.2). MFSIQ for children with mater- nally inherited deletions (63.7) was statistically lower D. E. McGinn1,2, M. Unolt3,4, T. B. Crowley1, B. S. Emanuel1,5, (p = .03) than for paternally inherited deletions (72.0). As E. H. Zackai1,5, E. Moss1, B. Morrow6, B. Nowakowska7,J. compared with the NY cohort where the MFSIQ for Vermeesch8, A. -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7, -

Ion Channels: Function Unravelled by Dysfunction
REVIEW ocus Ion channels: Function unravelled by dysfunction Thomas J. Jentsch, Christian A. Hübner and Jens C. Fuhrmann Ion channels allow the passage of specific ions and electrical charge. Plasma membrane channels are, for example, important for electrical excitability and transepithelial transport, whereas intracellular channels have roles in acidifying endosomes or in releasing Ca2+ from stores. The function of several channels emerged from mutations in humans or mice. The resulting phenotypes include kidney stones resulting from impaired endocytosis, hypertension, defective insulin secretion, cardiac arrhythmias, neurological diseases like epilepsy or deafness and even ‘developmental’ defects such as osteopetrosis. Ion channels are integral membrane proteins that form a pore to allow the of the selective expression of ion channels and transporters in apical passage of specific ions by passive diffusion. Most, if not all, ion channels or basolateral membranes. Typically, the ion is transported across one undergo conformational changes from closed to open states, and once of these membrane domains in an active transport process: primary open, channels allow the passage of thousands of ions. This distinguishes active transport occurs through transport ATPases (for example, the them from transporters and pumps, which can also transport ions but (Na+ + K+)ATPase), and secondary active transport couples trans- only one (or a few) at a time. The opening and closing of channels can be membrane ion gradients established by primary active transport (for controlled by various means, including voltage, the binding of ligands such example, the Na+ gradient) to the transport of another ion (for example, as intracellular Ca2+ or extracellular neurotransmitters, and post-transla- Cl– in Na–Cl cotransporters). -

Connexins and Disease
Downloaded from http://cshperspectives.cshlp.org/ on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Connexins and Disease Mario Delmar,1 Dale W. Laird,2 Christian C. Naus,3 Morten S. Nielsen,4 Vytautas K. Verselis,5 and Thomas W. White6 1The Leon H. Charney Division of Cardiology, New York University School of Medicine, New York, New York 10016 2Department of Anatomy and Cell Biology, University of Western Ontario, London, Ontario N6A5C1, Canada 3Department of Cellular and Physiological Sciences, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada 4Department of Biological Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen 2200, Denmark 5Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, New York, New York 10461 6Department of Physiology and Biophysics, Stony Brook University, Stony Brook, New York 11790 Correspondence: [email protected] Inherited or acquired alterations in the structure and function of connexin proteins have long been associated with disease. In the present work, we review current knowledge on the role of connexins in diseases associated with the heart, nervous system, cochlea, and skin, as well as cancer and pleiotropic syndromes such as oculodentodigital dysplasia (ODDD). Although incomplete by virtue of space and the extent of the topic, this review emphasizes the fact that connexin function is not only associated with gap junction channel formation. As such, both canonical and noncanonical functions of connexins are fundamental components in the pathophysiology of multiple connexin related disorders, many of them highly debilitating and life threatening. Improved understanding of connexin biology has the potential to advance our understanding of mechanisms, diagnosis, and treatment of disease. -
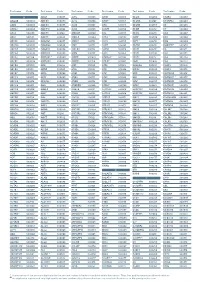
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

Wo2019/135816
( (51) International Patent Classification: GOPADHYAY, Soumyashree Ashok [US/US]; c/o 75 A61K 48/00 (2006.01) Francis Street, Boston, Massachusetts 021 15 (US). (21) International Application Number: (74) Agent: RUTLEDGE, Rachel D. et al.; Johnson, Marcou & PCT/US20 18/057 182 Isaacs, LLC, P. O .Box 691, Floschton, Georgia 30548 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 23 October 2018 (23. 10.2018) kind of national protection av ailable) . AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, (25) Filing Language: English CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (26) Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, (30) Priority Data: KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, 62/575,948 23 October 2017 (23. 10.2017) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 62/765,347 20 August 2018 (20.08.2018) US OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (71) Applicants: THE BROAD INSTITUTE, INC. [US/US]; SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 415 Main Street, Cambridge, Massachusetts 02142 (US). TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. THE BRIGHAM AND WOMEN'S HOSPITAL, INC. (84) Designated States (unless otherwise indicated, for every [US/US]; 75 Francis Street, Boston, Massachusetts 021 15 kind of regional protection available) . -

Murine Perinatal Beta Cell Proliferation and the Differentiation of Human Stem Cell Derived Insulin Expressing Cells Require NEUROD1
Page 1 of 105 Diabetes Murine perinatal beta cell proliferation and the differentiation of human stem cell derived insulin expressing cells require NEUROD1 Anthony I. Romer,1,2 Ruth A. Singer1,3, Lina Sui2, Dieter Egli,2* and Lori Sussel1,4* 1Department of Genetics and Development, Columbia University, New York, NY 10032, USA 2Department of Pediatrics, Columbia University, New York, NY 10032, USA 3Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, NY 10032, USA 4Department of Pediatrics, University of Colorado Denver School of Medicine, Denver, CO 80045, USA *Co-Corresponding Authors Dieter Egli 1150 St. Nicholas Avenue New York, NY 10032 [email protected] Lori Sussel 1775 Aurora Ct. Aurora, CO 80045 [email protected] Word Count: Abstract= 149; Body= 4773 Total Paper Figures= 7, Total Supplemental Tables= 4, Total Supplemental Figures= 5 Diabetes Publish Ahead of Print, published online September 13, 2019 Diabetes Page 2 of 105 Abstract Inactivation of the β cell transcription factor NEUROD1 causes diabetes in mice and humans. In this study, we uncovered novel functions of Neurod1 during murine islet cell development and during the differentiation of human embryonic stem cells (HESCs) into insulin-producing cells. In mice, we determined that Neurod1 is required for perinatal proliferation of alpha and beta cells. Surprisingly, apoptosis only makes a minor contribution to beta cell loss when Neurod1 is deleted. Inactivation of NEUROD1 in HESCs severely impaired their differentiation from pancreatic progenitors into insulin expressing (HESC-beta) cells; however survival or proliferation was not affected at the time points analyzed. NEUROD1 was also required in HESC-beta cells for the full activation of an essential beta cell transcription factor network.