Downloaded from Genbank (Using the Accession Numbers in the GTDB) to Perform a Comparative Genomic Analysis in Anvi’O Version 5.5 (Eren Et Al., 2015)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bacterial Communities of the Upper Respiratory Tract of Turkeys
www.nature.com/scientificreports OPEN Bacterial communities of the upper respiratory tract of turkeys Olimpia Kursa1*, Grzegorz Tomczyk1, Anna Sawicka‑Durkalec1, Aleksandra Giza2 & Magdalena Słomiany‑Szwarc2 The respiratory tracts of turkeys play important roles in the overall health and performance of the birds. Understanding the bacterial communities present in the respiratory tracts of turkeys can be helpful to better understand the interactions between commensal or symbiotic microorganisms and other pathogenic bacteria or viral infections. The aim of this study was the characterization of the bacterial communities of upper respiratory tracks in commercial turkeys using NGS sequencing by the amplifcation of 16S rRNA gene with primers designed for hypervariable regions V3 and V4 (MiSeq, Illumina). From 10 phyla identifed in upper respiratory tract in turkeys, the most dominated phyla were Firmicutes and Proteobacteria. Diferences in composition of bacterial diversity were found at the family and genus level. At the genus level, the turkey sequences present in respiratory tract represent 144 established bacteria. Several respiratory pathogens that contribute to the development of infections in the respiratory system of birds were identifed, including the presence of Ornithobacterium and Mycoplasma OTUs. These results obtained in this study supply information about bacterial composition and diversity of the turkey upper respiratory tract. Knowledge about bacteria present in the respiratory tract and the roles they can play in infections can be useful in controlling, diagnosing and treating commercial turkey focks. Next-generation sequencing has resulted in a marked increase in culture-independent studies characterizing the microbiome of humans and animals1–6. Much of these works have been focused on the gut microbiome of humans and other production animals 7–11. -

The Crystal Structure of the N-Acetylglucosamine 2-Epimerase from Nostoc Sp
research papers The crystal structure of the N-acetylglucosamine 2-epimerase from Nostoc sp. KVJ10 reveals the true dimer ISSN 2059-7983 Marie-Jose´e Haglund Halsør, Ulli Rothweiler, Bjørn Altermark and Inger Lin Uttakleiv Raeder* Received 26 September 2018 The Norwegian Structural Biology Centre (NorStruct), Department of Chemistry, UiT – The Arctic University of Norway, Accepted 30 November 2018 9037 Tromsø, Norway. *Correspondence e-mail: [email protected] Edited by M. Czjzek, Station Biologique de N-Acetylglucosamine 2-epimerases (AGEs) catalyze the interconversion of Roscoff, France N-acetylglucosamine and N-acetylmannosamine. They can be used to perform the first step in the synthesis of sialic acid from N-acetylglucosamine, which Keywords: N-acetylglucosamine 2-epimerase; makes the need for efficient AGEs a priority. This study presents the structure of AGE; sialic acid; crystal packing; ManNAc; the AGE from Nostoc sp. KVJ10 collected in northern Norway, referred to as GlcNAc; N-acetylmannosamine; Nostoc sp. nAGE10. It is the third AGE structure to be published to date, and the first one KVJ10. in space group P42212. The nAGE10 monomer folds as an ( / )6 barrel in a PDB reference: N-acetylglucosamine similar manner to that of the previously published AGEs, but the crystal did not 2-epimerase, 6f04 contain the dimers that have previously been reported. The previously proposed ‘back-to-back’ assembly involved the face of the AGE monomer where the Supporting information: this article has barrel helices are connected by small loops. Instead, a ‘front-to-front’ dimer was supporting information at journals.iucr.org/d found in nAGE10 involving the long loops that connect the barrel helices at this end. -

Cellobiose 2-Epimerase, Process for Producing
(19) TZZ ¥ZZ_T (11) EP 2 395 080 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/00 (2006.01) C12N 1/15 (2006.01) 06.08.2014 Bulletin 2014/32 C12N 1/19 (2006.01) C12N 1/21 (2006.01) C12N 5/10 (2006.01) C12N 9/90 (2006.01) (2006.01) (2006.01) (21) Application number: 10738433.1 C12N 15/09 C12P 19/00 (22) Date of filing: 25.01.2010 (86) International application number: PCT/JP2010/050928 (87) International publication number: WO 2010/090095 (12.08.2010 Gazette 2010/32) (54) CELLOBIOSE 2-EPIMERASE, PROCESS FOR PRODUCING SAME, AND USE OF SAME CELLOBIOSE 2-EPIMERASE, HERSTELLUNGSVERFAHREN DAFÜR UND VERWENDUNG CELLOBIOSE 2-ÉPIMÉRASE, PROCÉDÉ DE PRODUCTION DE CELLE-CI ET UTILISATION DE CELLE-CI (84) Designated Contracting States: (74) Representative: Daniels, Jeffrey Nicholas AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Page White & Farrer HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL Bedford House PT RO SE SI SK SM TR John Street London WC1N 2BF (GB) (30) Priority: 05.02.2009 JP 2009025070 (56) References cited: (43) Date of publication of application: WO-A1-2008/062555 14.12.2011 Bulletin 2011/50 • PARK CHANG-SU ET AL: "Characterization of a (73) Proprietor: Hayashibara Co., Ltd. recombinant cellobiose 2-epimerase from Okayama-shi, Okayama (JP) Caldicellulosiruptor saccharolyticus and its application in the production of mannose from (72) Inventors: glucose.", APPLIED MICROBIOLOGY AND • WATANABE Hikaru BIOTECHNOLOGY DEC 2011 LNKD- PUBMED: Okayama-shi 21691788,vol. -

Identification of Pasteurella Species and Morphologically Similar Organisms
UK Standards for Microbiology Investigations Identification of Pasteurella species and Morphologically Similar Organisms Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 1 of 28 © Crown copyright 2015 Identification of Pasteurella species and Morphologically Similar Organisms Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 2 of 28 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Pasteurella species and Morphologically Similar Organisms Contents ACKNOWLEDGMENTS ......................................................................................................... -

HACEK Endocarditis: State-Of-The-Art Matthieu Revest, Gérald Egmann, Vincent Cattoir, Pierre Tattevin
HACEK endocarditis: state-of-the-art Matthieu Revest, Gérald Egmann, Vincent Cattoir, Pierre Tattevin To cite this version: Matthieu Revest, Gérald Egmann, Vincent Cattoir, Pierre Tattevin. HACEK endocarditis: state- of-the-art. Expert Review of Anti-infective Therapy, Expert Reviews, 2016, 14 (5), pp.523-530. 10.1586/14787210.2016.1164032. hal-01296779 HAL Id: hal-01296779 https://hal-univ-rennes1.archives-ouvertes.fr/hal-01296779 Submitted on 10 Jun 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. HACEK endocarditis: state-of-the-art Matthieu Revest1, Gérald Egmann2, Vincent Cattoir3, and Pierre Tattevin†1 ¹Infectious Diseases and Intensive Care Unit, Pontchaillou University Hospital, Rennes; ²Department of Emergency Medicine, SAMU 97.3, Centre Hospitalier Andrée Rosemon, Cayenne; 3Bacteriology, Pontchaillou University Hospital, Rennes, France †Author for correspondence: Prof. Pierre Tattevin, Infectious Diseases and Intensive Care Unit, Pontchaillou University Hospital, 2, rue Henri Le Guilloux, 35033 Rennes Cedex 9, France Tel.: +33 299289564 Fax.: + 33 299282452 [email protected] Abstract The HACEK group of bacteria – Haemophilus parainfluenzae, Aggregatibacter spp. (A. actinomycetemcomitans, A. aphrophilus, A. paraphrophilus, and A. segnis), Cardiobacterium spp. (C. hominis, C. valvarum), Eikenella corrodens, and Kingella spp. -

Product Sheet Info
Product Information Sheet for HM-206 Aggregatibacter aphrophilus, Oral Taxon immediately upon arrival. For long-term storage, the vapor phase of a liquid nitrogen freezer is recommended. Freeze- 545, Strain F0387 thaw cycles should be avoided. Catalog No. HM-206 Growth Conditions: Media: For research use only. Not for human use. Haemophilus Test medium or equivalent Chocolate agar or equivalent Contributor: Incubation: Jacques Izard, Assistant Member of the Staff, Department of Temperature: 37°C Molecular Genetics, The Forsyth Institute, Boston, Atmosphere: Aerobic with 5% CO2 Massachusetts, USA Propagation: 1. Keep vial frozen until ready for use, then thaw. Manufacturer: 2. Transfer the entire thawed aliquot into a single tube of broth. BEI Resources 3. Use several drops of the suspension to inoculate an agar slant and/or plate. Product Description: 4. Incubate the tube, slant and/or plate at 37°C for 24 to Bacteria Classification: Pasteurellaceae, Aggregatibacter 48 hours. Species: Aggregatibacter aphrophilus (formerly Haemophilus 1 aphrophilus) Citation: Subtaxon: Oral Taxon 545 Acknowledgment for publications should read “The following Strain: F0387 reagent was obtained through BEI Resources, NIAID, NIH as Original Source: Aggregatibacter aphrophilus (A. part of the Human Microbiome Project: Aggregatibacter aphrophilus), Oral Taxon 545, strain F0387 was isolated in aphrophilus, Oral Taxon 545, Strain F0387, HM-206.” 1984 from the subgingival dental plaque, at a healthy site, 2,3 of a 24-year-old female patient in the United States. Comments: A. aphrophilus, Oral Taxon 545, strain F0387 Biosafety Level: 1 (HMP ID 9335) is a reference genome for The Human Appropriate safety procedures should always be used with Microbiome Project (HMP). -
![Haemophilus] Haemoglobinophilus As Canicola Haemoglobinophilus Gen](https://docslib.b-cdn.net/cover/6465/haemophilus-haemoglobinophilus-as-canicola-haemoglobinophilus-gen-1246465.webp)
Haemophilus] Haemoglobinophilus As Canicola Haemoglobinophilus Gen
Scotland's Rural College Reclassification of [Haemophilus] haemoglobinophilus as Canicola haemoglobinophilus gen. nov., comb. nov. including Bisgaard taxon 35 Christensen, Henrik; Kuhnert, Peter; Foster, Geoffrey; Bisgaard, Magne Published in: International Journal of Systematic and Evolutionary Microbiology DOI: 10.1099/ijsem.0.004881 First published: 15/07/2021 Document Version Peer reviewed version Link to publication Citation for pulished version (APA): Christensen, H., Kuhnert, P., Foster, G., & Bisgaard, M. (2021). Reclassification of [Haemophilus] haemoglobinophilus as Canicola haemoglobinophilus gen. nov., comb. nov. including Bisgaard taxon 35. International Journal of Systematic and Evolutionary Microbiology, 71(7), [004881]. https://doi.org/10.1099/ijsem.0.004881 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 27. Sep. 2021 1 Supplemetary material for the paper: 2 Reclassification of [Haemophilus] haemoglobinophilus as Canicola haemoglobinophilus 3 gen. nov., comb. nov. including Bisgaard taxon 35 4 By Henrik Christensen, Peter Kuhnert, Geoff Foster and Magne Bisgaard 5 1 Table S1. -

Pasteurellaceae: P. Multocida, Avibacterium Gallinarum, A
Pasteurellaceae: P. Multocida, Avibacterium gallinarum, A. paragallinarum All the members of the family Pasteurellaceae are gram negative coccobacilli. They are facultative anaerobes, and typically oxidase-positive (which sets them apart from members of the family Enterobacteriaceae). Morphology and Staining Members of the genera Avibacterium, and Pasteurella are gram-negative coccobacilli. Bipolarity, that is, the staining of only the tips of cells, may be demonstrable with polychrome stains (e.g., Wright’s stain). Cell structure Adhesins. Some and probably all members of the family Pasteurellaceae produce adhesins (and possibly more than one kind). A type 4 fimbria (adhesin) has been described for avian strains of P. Multocida. Capsule. The hyaluronic acid capsule of type A strains P. multocida serves as an adhesin. The hyaluronic acid is similar (if not identical) to host tissue components, and is thus poorly antigenic; they also bind complement components poorly (and is therefore antiphagocytic).The hyaluronic acid capsule also serves as an adhesin for respiratory tract epithelial cells as in the case of capsule type A strains of P. Multocida Exotoxin. Pasteurella produce a number of proteins with toxic activity. At least two of these are important in the pathogenesis of disease: RTX and Rho toxin Growth Characteristics Avibacterium and Pasteurella grow best in the presence of serum or blood. After overnight incubation (35–37 ◦C), colonies are up to 2 mm in diameter, clear to 1 grayish, and smooth or mucoid. All are gram-negative, nonmotile coccobacilli. They are facultative anaerobes, typically oxidase-positive. Variability P. multocida consists of 5 capsular serogroups (A, B, D, E, and F) and 16 somatic serotypes (1–16). -

Actinomyces Naeslundii and Aggregatibacter Aphrophilus Brain Abscess in an Adolescent
Arch Clin Med Case Rep 2019; 3 (6): 409-413 DOI: 10.26502/acmcr.96550112 Case Report Actinomyces Naeslundii and Aggregatibacter Aphrophilus Brain Abscess in an Adolescent Michael Croix1, Christopher Schwarz2, Ryan Breuer3,4, Amanda B. Hassinger3,4, Kunal Chadha5, Mark Daniel Hicar4,6 1Division of Internal Medicine and Pediatrics, University at Buffalo. Buffalo, New York, USA 2Division of Emergency Medicine, University at Buffalo. Buffalo, New York, USA 3Division of Pediatric Critical Care, John R. Oishei Children’s Hospital. Buffalo, New York, USA 4Department of Pediatrics, University at Buffalo. Buffalo, New York, USA 5Division of Pediatric Emergency Medicine, University at Buffalo. Buffalo, New York, USA 6Division of Pediatric Infectious Diseases, University at Buffalo. Buffalo, New York, USA *Corresponding Authors: Dr. Mark Daniel Hicar, Department of Pediatrics, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, 1001 Main Street, Buffalo, NY, 14203 USA, Tel: (716) 323-0150; Fax: (716) 888-3804; E-mail: [email protected] (or) [email protected] Dr. Michael Croix, Division of Internal Medicine and Pediatrics, 300 Linwood Ave, Buffalo, NY, 14209 USA, Tel: (217) 840-5750; Fax: (716) 888-3804; E-mail: [email protected] Received: 20 July 2019; Accepted: 02 August 2019; Published: 04 November 2019 Abstract We report the case of a child with a brain abscess from which Actinomyces naeslundii and Aggregatibacter aphrophilus were isolated. The is the first case describing A. naeslundii causing a brain abscess. This case highlights the association of these two organisms which may affect antibiotic choice and therapy length. Keywords: Brain; Abscess; Actinomyces; Aggregatibacter 1. Case Report A 13 year old male with no known past medical history initially presented to the Emergency Department with one week of headache, nausea, and vomiting. -
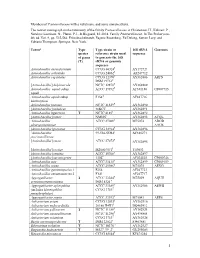
Members of Pasteurellaceae with a Valid Name and Some Unnamed Taxa
Members of Pasteurellaceae with a valid name and some unnamed taxa. The newest monograph on the taxonomy of the family Pasteurellaceae is Christensen, H., Kuhnert, P., Nørskov-Lauritsen, N., Planet, P.J., & Bisgaard, M. 2014. Family Pasteurellaceae. In The Prokaryotes 4th ed. Vol. 9, pp. 535-564. Erko Stackebrandt, Eugene Rosenberg, Ed Delong, Steven Lory, and Fabiano Thompson. Springer, New York. Taxona Type Type strain or 16S rRNA Genomes species reference strain used sequence of genus to generate the 16S (T) rRNA or genomic sequence Actinobacillus anseriformium CCUG 60324T AY172727 Actinobacillus arthritidis CCUG 24862T AF247712 Actinobacillus capsulatus CCUG 12396T AY362886 ARFN DSM 19761T [Actinobacillus] delphinicola NCTC 12870T AY362889 Actinobacillus equuli subsp. ATCC 19392T AF381186 CP007715 equuli Actinobacillus equuli subsp. F154T AF247716 haemolyticus Actinobacillus hominis NCTC 11529T AY362890 [Actinobacillus] indolicus 46KC2T AY362891 Actinobacillus lignieresii T NCTC 4189T AY362892 [Actinobacillus] minor NM305T AY362893 ACQL Actinobacillus ATCC 27088T M75074 ADOD pleuropneumoniae AACK [Actinobacillus] porcinus CCUG 38924T AY362896 ‘Actinobacillus 99-536-55H-F AF486274 porcitonsillarum’ T [Actinobacillus] rossii ATCC 27072 AY362895 [Actinobacillus] scotiae M2000/95/1T Y09653 [Actinobacillus] seminis ATCC 15768T AY362897 [Actinobacillus] succinogenes 130ZT AF024525 CP000746 Actinobacillus suis ATCC 33415T AY362899 CP009159 Actinobacillus ureae ATCC 25986T M75075 AEVG Actinobacillus genomospecies 1 F264 AF247723 Actinobacillus -

Phylogenomic and Molecular Demarcation of the Core Members of the Polyphyletic Pasteurellaceae Genera Actinobacillus, Haemophilus,Andpasteurella
Hindawi Publishing Corporation International Journal of Genomics Volume 2015, Article ID 198560, 15 pages http://dx.doi.org/10.1155/2015/198560 Research Article Phylogenomic and Molecular Demarcation of the Core Members of the Polyphyletic Pasteurellaceae Genera Actinobacillus, Haemophilus,andPasteurella Sohail Naushad, Mobolaji Adeolu, Nisha Goel, Bijendra Khadka, Aqeel Al-Dahwi, and Radhey S. Gupta Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON, Canada L8N 3Z5 Correspondence should be addressed to Radhey S. Gupta; [email protected] Received 5 November 2014; Revised 19 January 2015; Accepted 26 January 2015 Academic Editor: John Parkinson Copyright © 2015 Sohail Naushad et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The genera Actinobacillus, Haemophilus, and Pasteurella exhibit extensive polyphyletic branching in phylogenetic trees and do not represent coherent clusters of species. In this study, we have utilized molecular signatures identified through comparative genomic analyses in conjunction with genome based and multilocus sequence based phylogenetic analyses to clarify the phylogenetic and taxonomic boundary of these genera. We have identified large clusters of Actinobacillus, Haemophilus, and Pasteurella species which represent the “sensu stricto” members of these genera. We have identified 3, 7, and 6 conserved signature indels (CSIs), which are specifically shared by sensu stricto members of Actinobacillus, Haemophilus, and Pasteurella, respectively. We have also identified two different sets of CSIs that are unique characteristics of the pathogen containing genera Aggregatibacter and Mannheimia, respectively. It is now possible to demarcate the genera Actinobacillus sensu stricto, Haemophilus sensu stricto, and Pasteurella sensu stricto on the basis of discrete molecular signatures. -

Along the Path of Bacterial Nonulosonic Acids
Faculty of Science and Technology Along the path of bacterial nonulosonic acids A study of the bio- and in vitro synthesis of sialic acid related compounds — Marie-Josée Haglund Halsør A dissertation for the degree of Philosophiae Doctor – June 2019 Along the path of nonulosonic acids A study of the bio- and in vitro synthesis of sialic acid related compounds Marie-Josée Haglund Halsør A dissertation for the degree of Philosophiae Doctor FACULTY OF SCIENCE AND TECHNOLOGY DEPARTMENT OF CHEMISTRY June 2019 "There is a single light of science and to brighten it anywhere is to brighten it everywhere." - Unsourced, credited to Isaac Asimov. Preface “Why?”, and later “How?”. Those two questions are what led me to research, without doubt. I’ve asked them (aloud or not) every day for as long as I can remember, about practically everything. The other thing is being amazed by Nature. The diversity of every aspect and how it all functions as one, somehow. My favorite as a child were the documentaries by “le Commandant Cousteau” (the sharks!), and my dream was to be an oceanographer. I pursued that dream up until my first year of university, when I discovered biochemistry. I had already grown a liking for chemistry, and it was the only discipline that answered the “biological whys and hows” without going into physics. Biochemistry studies and does, both trying to unravel Nature’s secrets and building its own means to do so. It also uses the knowledge to improve human living conditions, at least in theory. I was sold, and here I am.