A Genomic Analysis Pipeline and Its Application to Pediatric Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-Rnns and the Protein Structure Prediction Problem
Journal of Machine Learning Research 4 (2003) 575-602 Submitted 2/02; Revised 4/03; Published 9/03 The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-RNNs and the Protein Structure Prediction Problem Pierre Baldi [email protected] Gianluca Pollastri [email protected] School of Information and Computer Science Institute for Genomics and Bioinformatics University of California, Irvine Irvine, CA 92697-3425, USA Editor: Michael I. Jordan Abstract We describe a general methodology for the design of large-scale recursive neural network architec- tures (DAG-RNNs) which comprises three fundamental steps: (1) representation of a given domain using suitable directed acyclic graphs (DAGs) to connect visible and hidden node variables; (2) parameterization of the relationship between each variable and its parent variables by feedforward neural networks; and (3) application of weight-sharing within appropriate subsets of DAG connec- tions to capture stationarity and control model complexity. Here we use these principles to derive several specific classes of DAG-RNN architectures based on lattices, trees, and other structured graphs. These architectures can process a wide range of data structures with variable sizes and dimensions. While the overall resulting models remain probabilistic, the internal deterministic dy- namics allows efficient propagation of information, as well as training by gradient descent, in order to tackle large-scale problems. These methods are used here to derive state-of-the-art predictors for protein structural features such as secondary structure (1D) and both fine- and coarse-grained contact maps (2D). Extensions, relationships to graphical models, and implications for the design of neural architectures are briefly discussed. -

Methodology for Predicting Semantic Annotations of Protein Sequences by Feature Extraction Derived of Statistical Contact Potentials and Continuous Wavelet Transform
Universidad Nacional de Colombia Sede Manizales Master’s Thesis Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform Author: Supervisor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez A thesis submitted in fulfillment of the requirements for the degree of Master’s on Engineering - Industrial Automation in the Department of Electronic, Electric Engineering and Computation Signal Processing and Recognition Group June 2014 Universidad Nacional de Colombia Sede Manizales Tesis de Maestr´ıa Metodolog´ıapara predecir la anotaci´on sem´antica de prote´ınaspor medio de extracci´on de caracter´ısticas derivadas de potenciales de contacto y transformada wavelet continua Autor: Tutor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez Tesis presentada en cumplimiento a los requerimientos necesarios para obtener el grado de Maestr´ıaen Ingenier´ıaen Automatizaci´onIndustrial en el Departamento de Ingenier´ıaEl´ectrica,Electr´onicay Computaci´on Grupo de Procesamiento Digital de Senales Enero 2014 UNIVERSIDAD NACIONAL DE COLOMBIA Abstract Faculty of Engineering and Architecture Department of Electronic, Electric Engineering and Computation Master’s on Engineering - Industrial Automation Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform by Gustavo Alonso Arango Argoty In this thesis, a method to predict semantic annotations of the proteins from its primary structure is proposed. The main contribution of this thesis lies in the implementation of a novel protein feature representation, which makes use of the pairwise statistical contact potentials describing the protein interactions and geometry at the atomic level. -

Deep Learning in Chemoinformatics Using Tensor Flow
UC Irvine UC Irvine Electronic Theses and Dissertations Title Deep Learning in Chemoinformatics using Tensor Flow Permalink https://escholarship.org/uc/item/963505w5 Author Jain, Akshay Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, IRVINE Deep Learning in Chemoinformatics using Tensor Flow THESIS submitted in partial satisfaction of the requirements for the degree of MASTER OF SCIENCE in Computer Science by Akshay Jain Thesis Committee: Professor Pierre Baldi, Chair Professor Cristina Videira Lopes Professor Eric Mjolsness 2017 c 2017 Akshay Jain DEDICATION To my family and friends. ii TABLE OF CONTENTS Page LIST OF FIGURES v LIST OF TABLES vi ACKNOWLEDGMENTS vii ABSTRACT OF THE THESIS viii 1 Introduction 1 1.1 QSAR Prediction Methods . .2 1.2 Deep Learning . .4 2 Artificial Neural Networks(ANN) 5 2.1 Artificial Neuron . .5 2.2 Activation Function . .7 2.3 Loss function . .8 2.4 Optimization . .8 3 Deep Recursive Architectures 10 3.1 Recurrent Neural Networks (RNN) . 10 3.2 Recursive Neural Networks . 11 3.3 Directed Acyclic Graph Recursive Neural Networks (DAG-RNN) . 11 4 UG-RNN for small molecules 14 4.1 DAG Generation . 16 4.2 Local Information Vector . 16 4.3 Contextual Vectors . 17 4.4 Activity Prediction . 17 4.5 UG-RNN With Contracted Rings (UG-RNN-CR) . 18 4.6 Example: UG-RNN Model of Propionic Acid . 20 5 Implementation 24 6 Data & Results 26 6.1 Aqueous Solubility Prediction . 26 6.2 Melting Point Prediction . 28 iii 7 Conclusions 30 Bibliography 32 A Source Code 37 A.1 UGRNN . -

Course Outline
Department of Computer Science and Software Engineering COMP 6811 Bioinformatics Algorithms (Reading Course) Fall 2019 Section AA Instructor: Gregory Butler Curriculum Description COMP 6811 Bioinformatics Algorithms (4 credits) The principal objectives of the course are to cover the major algorithms used in bioinformatics; sequence alignment, multiple sequence alignment, phylogeny; classi- fying patterns in sequences; secondary structure prediction; 3D structure prediction; analysis of gene expression data. This includes dynamic programming, machine learning, simulated annealing, and clustering algorithms. Algorithmic principles will be emphasized. A project is required. Outline of Topics The course will focus on algorithms for protein sequence analysis. It will not cover genome assembly, genome mapping, or gene recognition. • Background in Biology and Genomics • Sequence Alignment: Pairwise and Multiple • Representation of Protein Amino Acid Composition • Profile Hidden Markov Models • Specificity Determining Sites • Curation, Annotation, and Ontologies • Machine Learning: Secondary Structure, Signals, Subcellular Location • Protein Families, Phylogenomics, and Orthologous Groups • Profile-Based Alignments • Algorithms Based on k-mers 1 Texts | in Library D. Higgins and W. Taylor (editors). Bioinformatics: Sequence, Structure and Databanks, Oxford University Press, 2000. A. D. Baxevanis and B. F. F. Ouelette. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins, Wiley, 1998. Richard Durbin, Sean R. Eddy, Anders Krogh, -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Deep Learning in Science Pierre Baldi Frontmatter More Information
Cambridge University Press 978-1-108-84535-9 — Deep Learning in Science Pierre Baldi Frontmatter More Information Deep Learning in Science This is the first rigorous, self-contained treatment of the theory of deep learning. Starting with the foundations of the theory and building it up, this is essential reading for any scientists, instructors, and students interested in artificial intelligence and deep learning. It provides guidance on how to think about scientific questions, and leads readers through the history of the field and its fundamental connections to neuro- science. The author discusses many applications to beautiful problems in the natural sciences, in physics, chemistry, and biomedicine. Examples include the search for exotic particles and dark matter in experimental physics, the prediction of molecular properties and reaction outcomes in chemistry, and the prediction of protein structures and the diagnostic analysis of biomedical images in the natural sciences. The text is accompanied by a full set of exercises at different difficulty levels and encourages out-of-the-box thinking. Pierre Baldi is Distinguished Professor of Computer Science at University of California, Irvine. His main research interest is understanding intelligence in brains and machines. He has made seminal contributions to the theory of deep learning and its applications to the natural sciences, and has written four other books. © in this web service Cambridge University Press www.cambridge.org Cambridge University Press 978-1-108-84535-9 — Deep Learning in -

The Roles of Trim15 and UCHL3 in the Ubiquitin-Mediated Cell Cycle Regulation Katerina Jerabkova
The roles of Trim15 and UCHL3 in the ubiquitin-mediated cell cycle regulation Katerina Jerabkova To cite this version: Katerina Jerabkova. The roles of Trim15 and UCHL3 in the ubiquitin-mediated cell cycle regulation. Cellular Biology. Université de Strasbourg; Univerzita Karlova (Prague), 2019. English. NNT : 2019STRAJ034. tel-02884583 HAL Id: tel-02884583 https://tel.archives-ouvertes.fr/tel-02884583 Submitted on 30 Jun 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE STRASBOURG ÉCOLE DOCTORALE DES SCIENCES DE LA VIE ET DE LA SANTÉ IGBMC - CNRS UMR 7104 - Inserm U 1258 THÈSE présentée par : Kateřina JEŘÁBKOVÁ soutenue le : 09 Octobre 2019 pour obtenir le grade de : Docteur de l’université de Strasbourg Discipline/ Spécialité : Aspects moléculaires et cellulaires de la biologie Les rôles de Trim15 et UCHL3 dans la régulation, médiée par l’ubiquitine, du cycle cellulaire. The roles of Trim15 and UCHL3 in the ubiquitin-mediated cell cycle regulation. THÈSE dirigée par : Mme SUMARA Izabela PhD, Université de Strasbourg Mme CHAWENGSAKSOPHAK -

Bringing Folding Pathways Into Strand Pairing Prediction
Bringing folding pathways into strand pairing prediction Jieun Jeong1,2, Piotr Berman1, and Teresa Przytycka2 1 Computer Science and Engineering Department The Pennsylvania State University University Park, PA 16802 USA 2 National Center for Biotechnology Information US National Library of Medicine, National Institutes of Health Bethesda, MD 20894 email: [email protected], [email protected], [email protected] Abstract. The topology of β-sheets is defined by the pattern of hydrogen- bonded strand pairing. Therefore, predicting hydrogen bonded strand partners is a fundamental step towards predicting β-sheet topology. In this work we report a new strand pairing algorithm. Our algorithm at- tempts to mimic elements of the folding process. Namely, in addition to ensuring that the predicted hydrogen bonded strand pairs satisfy basic global consistency constraints, it takes into account hypothetical folding pathways. Consistently with this view, introducing hydrogen bonds be- tween a pair of strands changes the probabilities of forming other strand pairs. We demonstrate that this approach provides an improvement over previously proposed algorithms. 1 Introduction The prediction of protein structure from protein sequence is a long-held goal that would provide invaluable information regarding the function of individ- ual proteins and the evolution of protein families. The increasing amount of sequence and structure data, made it possible to decouple the structure predic- tion problem from the problem of modeling of protein folding process. Indeed, a significant progress has been achieved by bioinformatics approaches such as homology modeling, threading, and assembly from fragments [16]. At the same time, the fundamental problem of how actually a protein acquires its final folded state remains a subject of controversy. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -
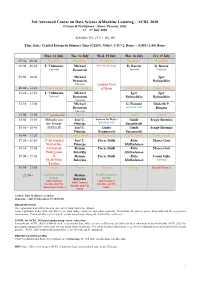
ACDL-2020-Programme-Ver10.0
3rd Advanced Course on Data Science &Machine Learning – ACDL 2020 Certosa di Pontignano - Siena, Tuscany, Italy 13 – 17 July 2020 Schedule Ver. 11.0 – July 9th Time Zone: Central European Summer Time (CEST), Offset: UTC+2, Rome – (GMT+2:00) Rome Mon, 13 July Tue, 14 July Wed, 15 July Thu, 16 July Fri, 17 July 07:30 – 09:00 Breakfast Breakfast Breakfast Breakfast Breakfast 09:00 – 09:50 T. Viehmann Michael 8:30 Social Tour D. Bacciu D. Bacciu Tutorial Bronstein Tutorial Tutorial 09:50 – 10:40 Michael Igor Bronstein Babuschkin Tutorial Guided Visit 10:40 – 11:20 Coffee break Coffee break of Siena Coffee break Coffee break 11:20 – 12:10 T. Viehmann Michael Igor Igor Tutorial Bronstein Babuschkin Babuschkin Tutorial 12:10 – 13:00 Michael G. Fiameni Diederik P. Bronstein Industrial Talk Kingma Tutorial 13:00 – 15:00 Lunch (13:00-14:00) Lunch Lunch Lunch Lunch 15:00 – 15:50 Mihaela van José C. Lorenzo De Mattei Guido Sergiy Butenko der Schaar Principe Industrial Talk Sanguinetti 15:50 – 16:40 (14:00-16:40) José C. Guido Guido Sergiy Butenko Principe Sanguinetti Sanguinetti 16:40 – 17:20 Coffee break Coffee break Coffee break Coffee break Coffee break 17:20 – 18:10 17:20 Guided José C. Pierre Baldi Risto Marco Gori Visit of the Principe Miikkulainen 18:10 – 19:00 Certosa di Roman Pierre Baldi Risto Marco Gori Pontignano Belavkin Miikkulainen 19:00 – 19:50 & Roman Pierre Baldi Risto Varun Ojha 18:20 Wine Belavkin Miikkulainen Tutorial Tasting 19:50 – 21:50 Dinner Dinner Dinner Dinner Social Dinner 21:50 – Oral Presentation Roman Oral Presentation Session Belavkin Session with Cantucci with Cantucci with Cantucci biscuits and Vin biscuits and Vin biscuits and Vin Santo (sweet wine) Santo Santo Arrival: July 12 (Dinner at 20:30) Departure: July 18 (Breakfast 07:30-09:00) REGISTRATION The registration desk will be located close to the Main Conference Room. -

I S C B N E W S L E T T
ISCB NEWSLETTER FOCUS ISSUE {contents} President’s Letter 2 Member Involvement Encouraged Register for ISMB 2002 3 Registration and Tutorial Update Host ISMB 2004 or 2005 3 David Baker 4 2002 Overton Prize Recipient Overton Endowment 4 ISMB 2002 Committees 4 ISMB 2002 Opportunities 5 Sponsor and Exhibitor Benefits Best Paper Award by SGI 5 ISMB 2002 SIGs 6 New Program for 2002 ISMB Goes Down Under 7 Planning Underway for 2003 Hot Jobs! Top Companies! 8 ISMB 2002 Job Fair ISCB Board Nominations 8 Bioinformatics Pioneers 9 ISMB 2002 Keynote Speakers Invited Editorial 10 Anna Tramontano: Bioinformatics in Europe Software Recommendations11 ISCB Software Statement volume 5. issue 2. summer 2002 Community Development 12 ISCB’s Regional Affiliates Program ISCB Staff Introduction 12 Fellowship Recipients 13 Awardees at RECOMB 2002 Events and Opportunities 14 Bioinformatics events world wide INTERNATIONAL SOCIETY FOR COMPUTATIONAL BIOLOGY A NOTE FROM ISCB PRESIDENT This newsletter is packed with information on development and dissemination of bioinfor- the ISMB2002 conference. With over 200 matics. Issues arise from recommendations paper submissions and over 500 poster submis- made by the Society’s committees, Board of sions, the conference promises to be a scientific Directors, and membership at large. Important feast. On behalf of the ISCB’s Directors, staff, issues are defined as motions and are discussed EXECUTIVE COMMITTEE and membership, I would like to thank the by the Board of Directors on a bi-monthly Philip E. Bourne, Ph.D., President organizing committee, local organizing com- teleconference. Motions that pass are enacted Michael Gribskov, Ph.D., mittee, and program committee for their hard by the Executive Committee which also serves Vice President work preparing for the conference. -

Pierre Baldi
Pierre Baldi School of Information and Computer Sciences Department of Computer Science Department of Biological Chemistry Institute for Genomics and Bioinformatics California Institute for Telecommunications and Information Technology University of California, Irvine Irvine, CA 92697-3435 (949) 824-5809 (949) 824-9813 FAX Assistant: Katarina Fletcher ([email protected]) [email protected] www.ics.uci.edu/~pfbaldi www.igb.uci.edu Professional Experience • October 2006 to present: Chancellor’s Professor • September 2006 to present: Associate-Director, Center for Machine Learning and Intelligent Systems. • January 2001 to present: Director Institute for Genomics and Bioinformatics. • June 2001 to present: Professor Department of Information and Computer Science, University of California, Irvine. [Joint appointment in the Department of Biological Chemistry, College of Medicine and the Department of Developmental and Cell Biology, School of Biological Sciences]. • January 2002 to December 2005: Application Layer Leader (Digitally Enabled Medicine) for the California Institute for Telecommunications and Information Technology [Calit2 2]. • July 1999 to May 2001: Associate Professor, Department of Information and Computer Science, University of California, Irvine. [Joint appointment in the Department of Biological Chemistry, College of Medicine and the Department of Developmental and Cell Biology, School of Biological Sciences]. • 1991 to June 1999: Chairman and CEO, Net-ID, Inc. • January 1999: Visiting Professor, Department of Computer Science, University of Florence. • 1995 to 1996: Member of the Professional Staff, Division of Biology, California Institute of Technology. • 1988 to 1995: Member of the Technical Staff in the Nonlinear Science and Information Processing Group at the Jet Propulsion Laboratory, and Visiting Research Associate, Division of Biology, California Institute of Technology.