Genetic Neuropathy Due to Impairments in Mitochondrial Dynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dynamin Functions and Ligands: Classical Mechanisms Behind
1521-0111/91/2/123–134$25.00 http://dx.doi.org/10.1124/mol.116.105064 MOLECULAR PHARMACOLOGY Mol Pharmacol 91:123–134, February 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics MINIREVIEW Dynamin Functions and Ligands: Classical Mechanisms Behind Mahaveer Singh, Hemant R. Jadhav, and Tanya Bhatt Department of Pharmacy, Birla Institute of Technology and Sciences Pilani, Pilani Campus, Rajasthan, India Received May 5, 2016; accepted November 17, 2016 Downloaded from ABSTRACT Dynamin is a GTPase that plays a vital role in clathrin-dependent pathophysiology of various disorders, such as Alzheimer’s disease, endocytosis and other vesicular trafficking processes by acting Parkinson’s disease, Huntington’s disease, Charcot-Marie-Tooth as a pair of molecular scissors for newly formed vesicles originating disease, heart failure, schizophrenia, epilepsy, cancer, dominant ’ from the plasma membrane. Dynamins and related proteins are optic atrophy, osteoporosis, and Down s syndrome. This review is molpharm.aspetjournals.org important components for the cleavage of clathrin-coated vesicles, an attempt to illustrate the dynamin-related mechanisms involved phagosomes, and mitochondria. These proteins help in organelle in the above-mentioned disorders and to help medicinal chemists division, viral resistance, and mitochondrial fusion/fission. Dys- to design novel dynamin ligands, which could be useful in the function and mutations in dynamin have been implicated in the treatment of dynamin-related disorders. Introduction GTP hydrolysis–dependent conformational change of GTPase dynamin assists in membrane fission, leading to the generation Dynamins were originally discovered in the brain and identi- of endocytic vesicles (Praefcke and McMahon, 2004; Ferguson at ASPET Journals on September 23, 2021 fied as microtubule binding partners. -

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

Targeted Genes and Methodology Details for Neuromuscular Genetic Panels
Targeted Genes and Methodology Details for Neuromuscular Genetic Panels Reference transcripts based on build GRCh37 (hg19) interrogated by Neuromuscular Genetic Panels Next-generation sequencing (NGS) and/or Sanger sequencing is performed Motor Neuron Disease Panel to test for the presence of a mutation in these genes. Gene GenBank Accession Number Regions of homology, high GC-rich content, and repetitive sequences may ALS2 NM_020919 not provide accurate sequence. Therefore, all reported alterations detected ANG NM_001145 by NGS are confirmed by an independent reference method based on laboratory developed criteria. However, this does not rule out the possibility CHMP2B NM_014043 of a false-negative result in these regions. ERBB4 NM_005235 Sanger sequencing is used to confirm alterations detected by NGS when FIG4 NM_014845 appropriate.(Unpublished Mayo method) FUS NM_004960 HNRNPA1 NM_031157 OPTN NM_021980 PFN1 NM_005022 SETX NM_015046 SIGMAR1 NM_005866 SOD1 NM_000454 SQSTM1 NM_003900 TARDBP NM_007375 UBQLN2 NM_013444 VAPB NM_004738 VCP NM_007126 ©2018 Mayo Foundation for Medical Education and Research Page 1 of 14 MC4091-83rev1018 Muscular Dystrophy Panel Muscular Dystrophy Panel Gene GenBank Accession Number Gene GenBank Accession Number ACTA1 NM_001100 LMNA NM_170707 ANO5 NM_213599 LPIN1 NM_145693 B3GALNT2 NM_152490 MATR3 NM_199189 B4GAT1 NM_006876 MYH2 NM_017534 BAG3 NM_004281 MYH7 NM_000257 BIN1 NM_139343 MYOT NM_006790 BVES NM_007073 NEB NM_004543 CAPN3 NM_000070 PLEC NM_000445 CAV3 NM_033337 POMGNT1 NM_017739 CAVIN1 NM_012232 POMGNT2 -
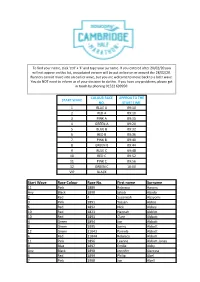
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Supplemental Material Table of Contents
Supplemental material Table of Contents Detailed Materials and Methods ......................................................................................................... 2 Perioperative period ........................................................................................................................... 2 Ethical aspects ................................................................................................................................... 4 Evaluation of heart failure ................................................................................................................. 4 Sample preparation for ANP mRNA expression .................................................................................. 5 Sample preparation for validative qRT-PCR (Postn, Myh7, Gpx3, Tgm2) ............................................ 6 Tissue fibrosis .................................................................................................................................... 7 Ventricular remodeling and histological tissue preservation ................................................................ 8 Evaluation of the histological preservation of cardiac tissue ................................................................ 9 Sample preparation and quantitative label-free proteomics analyses .................................................. 10 Statistical methods ........................................................................................................................... 12 References ........................................................................................................................................ -

Amyloid Β-Peptide Increases Mitochondria-Endoplasmic
cells Article Amyloid β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models Nuno Santos Leal 1,*, Giacomo Dentoni 1 , Bernadette Schreiner 1 , Luana Naia 1, Antonio Piras 1, Caroline Graff 1, Antonio Cattaneo 2, Giovanni Meli 2 , Maho Hamasaki 3, Per Nilsson 1 and Maria Ankarcrona 1,* 1 Division of Neurogeriatrics, Department of Neurobiology, Care Science and Society, Karolinska Institutet, BioClinicum J9:20, Visionsgatan 4, 171 64 Solna, Sweden; [email protected] (G.D.); [email protected] (B.S.); [email protected] (L.N.); [email protected] (A.P.); caroline.graff@ki.se (C.G.); [email protected] (P.N.) 2 European Brain Research Institute (EBRI), Viale Regina Elena 295, 00161 Roma, Italy; [email protected] (A.C.); [email protected] (G.M.) 3 Department of Genetics, Graduate School of Medicine, Osaka University, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan; [email protected] * Correspondence: [email protected] (N.S.L.); [email protected] (M.A.); Tel.: +44-122-333-4390 (N.S.L.); +46-852-483-577 (M.A.) Received: 23 October 2020; Accepted: 25 November 2020; Published: 28 November 2020 Abstract: Recent findings have shown that the connectivity and crosstalk between mitochondria and the endoplasmic reticulum (ER) at mitochondria–ER contact sites (MERCS) are altered in Alzheimer’s disease (AD) and in AD-related models. MERCS have been related to the initial steps of autophagosome formation as well as regulation of mitochondrial function. Here, the interplay between MERCS, mitochondria ultrastructure and function and autophagy were evaluated in different AD animal models with increased levels of Aβ as well as in primary neurons derived from these animals. -

Sézary Syndrome Is a Unique Cutaneous T-Cell Lymphoma As
Leukemia (2008) 22, 393–399 & 2008 Nature Publishing Group All rights reserved 0887-6924/08 $30.00 www.nature.com/leu ORIGINAL ARTICLE Se´zary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3 N Booken1,11, A Gratchev1,11, J Utikal1, C Wei2,XYu3, M Qadoumi1, M Schmuth4, N Sepp4, D Nashan5, K Rass6,TTu¨ting7, C Assaf8, E Dippel1,9, R Stadler10, C-D Klemke1 and S Goerdt1 1Department of Dermatology, Venereology and Allergology, University Medical Centre Mannheim, Ruprecht Karl University of Heidelberg, Mannheim, Germany; 2Institute of Medical Statistics, University Medical Centre Mannheim, Ruprecht Karl University of Heidelberg, Mannheim, Germany; 3Medical Research Center (ZMF), University Medical Centre Mannheim, Ruprecht Karl University of Heidelberg, Mannheim, Germany; 4Department of Dermatology, Innsbruck Medical University, Innsbruck, Austria; 5Department of Dermatology, University of Freiburg, Freiburg, Germany; 6Department of Dermatology, The Saarland University Hospital, Homburg/Saar, Germany; 7Department of Dermatology, University of Bonn, Bonn, Germany; 8Department of Dermatology, Charite-University Medicine Berlin, Berlin, Germany; 9Department of Dermatology, Academic Medical Centre, Lemgo, Germany and 10Department of Dermatology, Academic Medical Centre, Minden, Germany Sezary syndrome (SS) is a rare, aggressive CD4 þ cutaneous While MF usually is a slowly progressive disease, SS runs a more T-cell lymphoma (CTCL); molecular traits differentiating SS aggressive course with a high mortality rate and a median survival from nonleukemic mycosis fungoides (MF) and from inflamma- time of 2–4 years. The probability of survival in CTCL can be tory skin diseases (ID) are not sufficiently characterized. -

The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease
International Journal of Molecular Sciences Review The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease Chunling Huang * , Ji Bian , Qinghua Cao, Xin-Ming Chen and Carol A. Pollock * Kolling Institute, Sydney Medical School, Royal North Shore Hospital, University of Sydney, St. Leonards, NSW 2065, Australia; [email protected] (J.B.); [email protected] (Q.C.); [email protected] (X.-M.C.) * Correspondence: [email protected] (C.H.); [email protected] (C.A.P.); Tel.: +61-2-9926-4784 (C.H.); +61-2-9926-4652 (C.A.P.) Abstract: Mitochondria are critical organelles that play a key role in cellular metabolism, survival, and homeostasis. Mitochondrial dysfunction has been implicated in the pathogenesis of diabetic kidney disease. The function of mitochondria is critically regulated by several mitochondrial protein kinases, including the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1). The focus of PINK1 research has been centered on neuronal diseases. Recent studies have revealed a close link between PINK1 and many other diseases including kidney diseases. This review will provide a concise summary of PINK1 and its regulation of mitochondrial function in health and disease. The physiological role of PINK1 in the major cells involved in diabetic kidney disease including proximal tubular cells and podocytes will also be summarized. Collectively, these studies suggested that targeting PINK1 may offer a promising alternative for the treatment of diabetic kidney disease. Keywords: PINK1; diabetic kidney disease; mitochondria; mitochondria quality control; mitophagy Citation: Huang, C.; Bian, J.; Cao, Q.; 1. Introduction Chen, X.-M.; Pollock, C.A. -

Genome-Wide Analyses Identify KIF5A As a Novel ALS Gene
This is a repository copy of Genome-wide Analyses Identify KIF5A as a Novel ALS Gene.. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/129590/ Version: Accepted Version Article: Nicolas, A, Kenna, KP, Renton, AE et al. (210 more authors) (2018) Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron, 97 (6). 1268-1283.e6. https://doi.org/10.1016/j.neuron.2018.02.027 Reuse This article is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) licence. This licence only allows you to download this work and share it with others as long as you credit the authors, but you can’t change the article in any way or use it commercially. More information and the full terms of the licence here: https://creativecommons.org/licenses/ Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Genome-wide Analyses Identify KIF5A as a Novel ALS Gene Aude Nicolas1,2, Kevin P. Kenna2,3, Alan E. Renton2,4,5, Nicola Ticozzi2,6,7, Faraz Faghri2,8,9, Ruth Chia1,2, Janice A. Dominov10, Brendan J. Kenna3, Mike A. Nalls8,11, Pamela Keagle3, Alberto M. Rivera1, Wouter van Rheenen12, Natalie A. Murphy1, Joke J.F.A. van Vugt13, Joshua T. Geiger14, Rick A. Van der Spek13, Hannah A. Pliner1, Shankaracharya3, Bradley N. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers
Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers. by Kelly Kennaley A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Molecular and Cellular Pathology) in the University of Michigan 2019 Doctoral Committee: Associate Professor Zaneta Nikolovska-Coleska, Co-Chair Adjunct Associate Professor Scott A. Tomlins, Co-Chair Associate Professor Eric R. Fearon Associate Professor Alexey I. Nesvizhskii Kelly R. Kennaley [email protected] ORCID iD: 0000-0003-2439-9020 © Kelly R. Kennaley 2019 Acknowledgements I have immeasurable gratitude for the unwavering support and guidance I received throughout my dissertation. First and foremost, I would like to thank my thesis advisor and mentor Dr. Scott Tomlins for entrusting me with a challenging, interesting, and impactful project. He taught me how to drive a project forward through set-backs, ask the important questions, and always consider the impact of my work. I’m truly appreciative for his commitment to ensuring that I would get the most from my graduate education. I am also grateful to the many members of the Tomlins lab that made it the supportive, collaborative, and educational environment that it was. I would like to give special thanks to those I’ve worked closely with on this project, particularly Dr. Moloy Goswami for his mentorship, Lei Lucy Wang, Dr. Sumin Han, and undergraduate students Bhavneet Singh, Travis Weiss, and Myles Barlow. I am also grateful for the support of my thesis committee, Dr. Eric Fearon, Dr. Alexey Nesvizhskii, and my co-mentor Dr. Zaneta Nikolovska-Coleska, who have offered guidance and critical evaluation since project inception. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated.