Raman Spectroscopyspectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
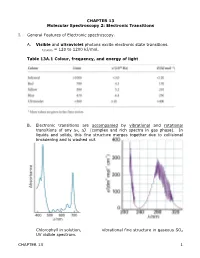
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. εphoton = 120 to 1200 kJ/mol. Table 13A.1 Colour, frequency, and energy of light B. Electronic transitions are accompanied by vibrational and rotational transitions of any Δν, ΔJ (complex and rich spectra in gas phase). In liquids and solids, this fine structure merges together due to collisional broadening and is washed out. Chlorophyll in solution, vibrational fine structure in gaseous SO2 UV visible spectrum. CHAPTER 13 1 C. Molecules need not have permanent dipole to undergo electronic transitions. All that is required is a redistribution of electronic and nuclear charge between the initial and final electronic state. Transition intensity ~ |µ|2 where µ is the magnitude of the transition dipole moment given by: ˆ d µ = ∫ ψfinalµψ initial τ where µˆ = −e ∑ri + e ∑Ziri € electrons nuclei r vector position of the particle i = D. Franck-Condon principle: Because nuclei are so much more massive and € sluggish than electrons, electronic transitions can happen much faster than the nuclei can respond. Electronic transitions occur vertically on energy diagram at right. (Hence the name vertical transition) Transition probability depends on vibrational wave function overlap (Franck-Condon factor). In this example: ν = 0 ν’ = 0 have small overlap ν = 0 ν’ = 2 have greatest overlap Now because the total molecular wavefunction can be approximately factored into 2 terms, electronic wavefunction and the nuclear position wavefunction (using Born-Oppenheimer approx.) the transition dipole can also be factored into 2 terms also: S µ = µelectronic i,f S d Franck Condon factor i,j i,f = ∫ ψf,vibr ψi,vibr τ = − CHAPTER 13 2 See derivation “Justification 14.2” II. -

Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO
Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO. WHEN we solve the Schrödinger equation, we always obtain two things: 1. a set of eigenstates, |ψn>. 2. a set of eigenstate energies, En QM predicts the existence of discrete, evenly spaced, vibrational energy levels for the SHO. n = 0,1,2,3... For the ground state (n=0), E = ½hν. Notes: This is called the zero point energy. Optical selection rule -- SHO can absorb or emit light with a ∆n = ±1 The IR absorption spectrum for a diatomic molecule, such as HCl: diatomic Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion. (Note: μ here means dipole moment). If a more realistic Morse potential is used in the Schrödinger Equation, these energy levels get scrunched together... ΔE=hν ΔE<hν The vibrational spectroscopy of polyatomic molecules gets more interesting... For diatomic or linear molecules: 3N-5 modes For nonlinear molecules: 3N-6 modes N = number of atoms in molecule The vibrational spectroscopy of polyatomic molecules gets more interesting... Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 All bands are observed in the IR spectrum. -

Lecture 17 Highlights
Lecture 23 Highlights The phenomenon of Rabi oscillations is very different from our everyday experience of how macroscopic objects (i.e. those made up of many atoms) absorb electromagnetic radiation. When illuminated with light (like French fries at McDonalds) objects tend to steadily absorb the light and heat up to a steady state temperature, showing no signs of oscillation with time. We did a calculation of the transition probability of an atom illuminated with a broad spectrum of light and found that the probability increased linearly with time, leading to a constant transition rate. This difference comes about because the many absorption processes at different frequencies produce a ‘smeared-out’ response that destroys the coherent Rabi oscillations and results in a classical incoherent absorption. Selection Rules All of the transition probabilities are proportional to “dipole matrix elements,” such as x jn above. These are similar to dipole moment calculations for a charge distribution in classical physics, except that they involve the charge distribution in two different states, connected by the dipole operator for the transition. In many cases these integrals are zero because of symmetries of the associated wavefunctions. This gives rise to selection rules for possible transitions under the dipole approximation (atom size much smaller than the electromagnetic wave wavelength). For example, consider the matrix element z between states of the n,l , m ; n ', l ', m ' Hydrogen atom labeled by the quantum numbers n,,l m and n',','l m : z = ψ()()r z ψ r d3 r n,l , m ; n ', l ', m ' ∫∫∫ n,,l m n',l ', m ' To make progress, first consider just the φ integral. -

Introduction to Differential Equations
Quantum Dynamics – Quick View Concepts of primary interest: The Time-Dependent Schrödinger Equation Probability Density and Mixed States Selection Rules Transition Rates: The Golden Rule Sample Problem Discussions: Tools of the Trade Appendix: Classical E-M Radiation POSSIBLE ADDITIONS: After qualitative section, do the two state system, and then first and second order transitions (follow Fitzpatrick). Chain together the dipole rules to get l = 2,0,-2 and m = -2, -2, … , 2. ??Where do we get magnetic rules? Look at the canonical momentum and the Asquared term. Schrödinger, Erwin (1887-1961) Austrian physicist who invented wave mechanics in 1926. Wave mechanics was a formulation of quantum mechanics independent of Heisenberg's matrix mechanics. Like matrix mechanics, wave mechanics mathematically described the behavior of electrons and atoms. The central equation of wave mechanics is now known as the Schrödinger equation. Solutions to the equation provide probability densities and energy levels of systems. The time-dependent form of the equation describes the dynamics of quantum systems. http://scienceworld.wolfram.com/biography/Schroedinger.html © 1996-2006 Eric W. Weisstein www-history.mcs.st-andrews.ac.uk/Biographies/Schrodinger.html Quantum Dynamics: A Qualitative Introduction Introductory quantum mechanics focuses on time-independent problems leaving Contact: [email protected] dynamics to be discussed in the second term. Energy eigenstates are characterized by probability density distributions that are time-independent (static). There are examples of time-dependent behavior that are by demonstrated by rather simple introductory problems. In the case of a particle in an infinite well with the range [ 0 < x < a], the mixed state below exhibits time-dependence. -

HJ Lipkin the Weizmann Institute of Science Rehovot, Israel
WHO UNDERSTANDS THE ZWEIG-IIZUKA RULE? H.J. Lipkin The Weizmann Institute of Science Rehovot, Israel Abstract: The theoretical basis of the Zweig-Iizuka Ru le is discussed with the emphasis on puzzles and paradoxes which remain open problems . Detailed analysis of ZI-violating transitions arising from successive ZI-allowed transitions shows that ideal mixing and additional degeneracies in the particle spectrum are required to enable cancellation of these higher order violations. A quantitative estimate of the observed ZI-violating f' + 2rr decay be second order transitions via the intermediate KK state agrees with experiment when only on-shell contributions are included . This suggests that ZI-violations for old particles are determined completely by on-shell contributions and cannot be used as input to estimate ZI-violation for new particle decays where no on-shell intermediate states exist . 327 INTRODUCTION - SOME BASIC QUESTIONS 1 The Zweig-Iizuka rule has entered the folklore of particle physics without any clear theoretical understanding or justification. At the present time nobody really understands it, and anyone who claims to should not be believed. Investigating the ZI rule for the old particles raises many interesting questions which may lead to a better understanding of strong interactions as well as giving additional insight into the experimentally observed suppression of new particle decays attributed to the ZI rule. This talk follows an iconoclastic approach emphasizing embarrassing questions with no simple answers which might lead to fruitful lines of investigation. Both theoretical and experimental questions were presented in the talk. However, the experimental side is covered in a recent paper2 and is not duplicated here. -

Surface-Enhanced Raman Scattering (SERS)
Focal Point Review Applied Spectroscopy 2018, Vol. 72(7) 987–1000 ! The Author(s) 2018 Surface-Enhanced Raman Scattering Reprints and permissions: sagepub.co.uk/journalsPermissions.nav (SERS) in Microbiology: Illumination and DOI: 10.1177/0003702818764672 journals.sagepub.com/home/asp Enhancement of the Microbial World Malama Chisanga, Howbeer Muhamadali, David I. Ellis, and Royston Goodacre Abstract The microbial world forms a huge family of organisms that exhibit the greatest phylogenetic diversity on Earth and thus colonize virtually our entire planet. Due to this diversity and subsequent complex interactions, the vast majority of microorganisms are involved in innumerable natural bioprocesses and contribute an absolutely vital role toward the maintenance of life on Earth, whilst a small minority cause various infectious diseases. The ever-increasing demand for environmental monitoring, sustainable ecosystems, food security, and improved healthcare systems drives the continuous search for inexpensive but reproducible, automated and portable techniques for detection of microbial isolates and understanding their interactions for clinical, environmental, and industrial applications and benefits. Surface-enhanced Raman scattering (SERS) is attracting significant attention for the accurate identification, discrimination and characteriza- tion and functional assessment of microbial cells at the single cell level. In this review, we briefly discuss the technological advances in Raman and Fourier transform infrared (FT-IR) instrumentation and their application for the analysis of clinically and industrially relevant microorganisms, biofilms, and biological warfare agents. In addition, we summarize the current trends and future prospects of integrating Raman/SERS-isotopic labeling and cell sorting technologies in parallel, to link genotype-to-phenotype in order to define community function of unculturable microbial cells in mixed microbial communities which possess admirable traits such as detoxification of pollutants and recycling of essential metals. -

UV-Vis. Molecular Absorption Spectroscopy
UV-Vis. Molecular Absorption Spectroscopy Prof. Tarek A. Fayed UV-Vis. Electronic Spectroscopy The interaction of molecules with ultraviolet and visible light may results in absorption of photons. This results in electronic transition, involving valance electrons, from ground state to higher electronic states (called excited states). The promoted electrons are electrons of the highest molecular orbitals HOMO. Absorption of ultraviolet and visible radiation in organic molecules is restricted to certain functional groups (known as chromophores) that contain valence electrons of low excitation energy. A chromophore is a chemical entity embedded within a molecule that absorbs radiation at the same wavelength in different molecules. Examples of Chromophores are dienes, aromatics, polyenes and conjugated ketones, etc. Types of electronic transitions Electronic transitions that can take place are of three types which can be considered as; Transitions involving p-, s-, and n-electrons. Transitions involving charge-transfer electrons. Transitions involving d- and f-electrons in metal complexes. Most absorption spectroscopy of organic molecules is based on transitions of n- or -electrons to the *-excited state. These transitions fall in an experimentally convenient region of the spectrum (200 - 700 nm), and need an unsaturated group in the molecule to provide the -electrons. In vacuum UV or far UV (λ<190 nm ) In UV/VIS For formaldehyde molecule; Selection Rules of electronic transitions Electronic transitions may be allowed or forbidden transitions, as reflected by appearance of an intense or weak band according to the magnitude of εmax, and is governed by the following selection rules : 1. Spin selection rule (△S = 0 for the transition to be allowed): there should be no change in spin orientation i. -
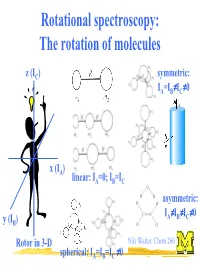
Rotational Spectroscopy: the Rotation of Molecules
Rotational spectroscopy: The rotation of molecules z (IC) symmetric: ≠ ≠ IA=IB IC 0 x (IA) linear: IA=0; IB=IC asymmetric: ≠ ≠ ≠ IA IB IC 0 y (IB) Rotor in 3-D Nils Walter: Chem 260 ≠ spherical: IA=IB=IC 0 The moment of inertia for a diatomic (linear) rigid rotor r definition: r = r1 + r2 m1 m2 C(enter of mass) r1 r2 Moment of inertia: Balancing equation: m1r1 = m2r2 2 2 I = m1r1 + m2r2 m m ⇒ I = 1 2 r 2 = µr 2 µ = reduced mass + m1 m2 Nils Walter: Chem 260 Solution of the Schrödinger equation for the rigid diatomic rotor h2 (No potential, − V 2Ψ = EΨ µ only kinetic 2 energy) ⇒ EJ = hBJ(J+1); rot. quantum number J = 0,1,2,... h B = [Hz] 4πI ⇒ EJ+1 -EJ = 2hB(J+1) Nils Walter: Chem 260 Selection rules for the diatomic rotor: 1. Gross selection rule Light is a transversal electromagnetic wave a polar rotor appears to have an oscillating ⇒ a molecule must be polar to electric dipole Nils Walter: Chem 260 be able to interact with light Selection rules for the diatomic rotor: 2. Specific selection rule rotational quantum number J = 0,1,2,… describes the angular momentum of a molecule (just like electronic orbital quantum number l=0,1,2,...) and light behaves as a particle: photons have a spin of 1, i.e., an angular momentum of one unit and the total angular momentum upon absorption or emission of a photon has to be preserved ⇒⇒∆∆J = ± 1 Nils Walter: Chem 260 Okay, I know you are dying for it: Schrödinger also can explain the ∆J = ± 1 selection rule Transition dipole µ = Ψ µΨ dτ moment fi ∫ f i initial state final state ⇒ only if this integral -

Complementary Vibrational Spectroscopy
Complementary Vibrational Spectroscopy Kazuki Hashimoto1,2, Venkata Ramaiah Badarla3, Akira Kawai1 and Takuro Ideguchi*3,4 1 Department of Physics, The University of Tokyo, Tokyo 113-0033, Japan 2 Aeronautical Technology Directorate, Japan Aerospace Exploration Agency, Tokyo 181-0015, Japan 3 Institute for Photon Science and Technology, The University of Tokyo, Tokyo 113-0033, Japan 4 PRESTO, Japan Science and Technology Agency, Saitama 332-0012, Japan *[email protected] Vibrational spectroscopy, comprised of infrared absorption and Raman scattering spectroscopy, is widely used for label-free optical sensing and imaging in various scientific and industrial fields. The group theory states that the two molecular spectroscopy methods are sensitive to vibrations categorized in different point groups and provide complementary vibrational spectra. Therefore, complete vibrational information cannot be acquired by a single spectroscopic device, which has impeded the full potential of vibrational spectroscopy. Here, we demonstrate simultaneous infrared absorption and Raman scattering spectroscopy that allows us to measure the complete broadband vibrational spectra in the molecular fingerprint region with a single instrument based on an ultrashort pulsed laser. The system is based on dual-modal Fourier-transform spectroscopy enabled by efficient use of nonlinear optical effects. Our proof-of-concept experiment demonstrates rapid, broadband and high spectral resolution measurements of complementary spectra of organic liquids for -

Shaping Polaritons to Reshape Selection Rules
Shaping Polaritons to Reshape Selection Rules 1,2 2 3 2 2,4 Francisco Machado∗ , Nicholas Rivera∗ , Hrvoje Buljan , Marin Soljaˇci´c , Ido Kaminer 1Department of Physics, University of California, Berkeley, CA 97420, USA 2Department of Physics, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 3Department of Physics, University of Zagreb, Zagreb 10000, Croatia. 4Department of Electrical Engineering, Technion, Israel Institute of Technology, Haifa 32000, Israel. The discovery of orbital angular momentum (OAM) in light established a new degree of freedom by which to control not only its flow but also its interaction with matter. Here, we show that by shaping extremely sub-wavelength polariton modes, for example by imbuing plasmon and phonon polariton with OAM, we engineer which transitions are allowed or forbidden in electronic systems such as atoms, molecules, and artificial atoms. Crucial to the feasibility of these engineered selection rules is the access to conventionally forbidden transitions afforded by sub-wavelength polaritons. We also find that the position of the absorbing atom provides a surprisingly rich parameter for controlling which absorption processes dominate over others. Additional tunability can be achieved by altering the polaritonic properties of the substrate, for example by tuning the carrier density in graphene, potentially enabling electronic control over selection rules. Our findings are best suited to OAM-carrying polaritonic modes that can be created in graphene, monolayer conductors, thin metallic films, and thin films of polar dielectrics such as boron nitride. By building on these findings we foresee the complete engineering of spectroscopic selection rules through the many degrees of freedom in the shape of optical fields. -

Infrared and Raman Spectroscopy UNIT II: Infrared and Raman Spectroscopy
CORE COURSE-VIII (CC-VIII) PHYSICAL METHODS IN CHEMISTRY II UNIT II: Infrared and Raman Spectroscopy UNIT II: Infrared and Raman Spectroscopy Vibrations in simple molecules (H2O, CO2) and their symmetry notation for molecular vibrations – group vibrations and the limitations – combined uses of IR and Raman spectroscopy in the - – structural elucidation of simple molecules like N2O, ClF3, NO3 , ClO4 effect of coordination on ligand vibrations – uses of groups vibrations in the structural elucidation of metal complexes of urea, thiourea, cyanide, thiocyanate and dimethyl sulfoxide. Effect of isotopic substitution on the vibrational spectra of molecules – vibrational spectra of metal carbonyls with reference to the nature of bonding – geometry and number of C-O stretching vibrations (group theoretical treatment) – applications of Raman spectroscopy – resonance Raman spectroscopy. Vibrations in simple molecules (H2O, CO2) Molecular Vibrations There are two types of molecular vibrations, stretching and bending. A molecule consisting of n atoms has a total of 3n degrees of freedom, corresponding to the Cartesian coordinates of each atom in the molecule. In a nonlinear molecule, 3 of these degrees are rotational and 3 are translational and the remaining corresponds to fundamental vibrations; in a linear molecule, 2 degrees are rotational and 3 are translational. The net number of fundamental vibrations for nonlinear and linear molecules is therefore: Molecule degrees of freedom: (nonlinear 3n– 6) (linear 3n– 5) The fundamental vibrations for water, H2O, are given in Figure. Water, which is nonlinear, has three fundamental vibrations. Carbon dioxide, CO2, is linear and hence has four fundamental vibrations (Figure). The two scissoring or bending vibrations are equivalent and therefore, have the same frequency and are said to be degenerate, appearing in an IR spectrum at 666 cm–1. -

Time Resolved Raman Spectroscopy for Depth Measurements Through
VU Research Portal Time-Resolved Raman Spectroscopy for depth analysis in scattering samples Petterson, I.E.I. 2013 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Petterson, I. E. I. (2013). Time-Resolved Raman Spectroscopy for depth analysis in scattering samples. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 30. Sep. 2021 Chapter 1. Introduction Concepts of Raman spectroscopy 11 1.1 Light scattering and the Raman effect Types of electromagnetic scattering When photons from a monochromatic light source such as a laser illuminate a material, the light can be transmitted, absorbed or scattered. In the case of scattering, the majority of this light is elastically scattered by the material with the same photon energy as the incident beam.