Gene Co-Expression Network Analysis for Identifying Modules and Functionally Enriched Pathways in SCA2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Information Proximity Interactions Among Centrosome
Current Biology, Volume 24 Supplemental Information Proximity Interactions among Centrosome Components Identify Regulators of Centriole Duplication Elif Nur Firat-Karalar, Navin Rauniyar, John R. Yates III, and Tim Stearns Figure S1 A Myc Streptavidin -tubulin Merge Myc Streptavidin -tubulin Merge BirA*-PLK4 BirA*-CEP63 BirA*- CEP192 BirA*- CEP152 - BirA*-CCDC67 BirA* CEP152 CPAP BirA*- B C Streptavidin PCM1 Merge Myc-BirA* -CEP63 PCM1 -tubulin Merge BirA*- CEP63 DMSO - BirA* CEP63 nocodazole BirA*- CCDC67 Figure S2 A GFP – + – + GFP-CEP152 + – + – Myc-CDK5RAP2 + + + + (225 kDa) Myc-CDK5RAP2 (216 kDa) GFP-CEP152 (27 kDa) GFP Input (5%) IP: GFP B GFP-CEP152 truncation proteins Inputs (5%) IP: GFP kDa 1-7481-10441-1290218-1654749-16541045-16541-7481-10441-1290218-1654749-16541045-1654 250- Myc-CDK5RAP2 150- 150- 100- 75- GFP-CEP152 Figure S3 A B CEP63 – – + – – + GFP CCDC14 KIAA0753 Centrosome + – – + – – GFP-CCDC14 CEP152 binding binding binding targeting – + – – + – GFP-KIAA0753 GFP-KIAA0753 (140 kDa) 1-496 N M C 150- 100- GFP-CCDC14 (115 kDa) 1-424 N M – 136-496 M C – 50- CEP63 (63 kDa) 1-135 N – 37- GFP (27 kDa) 136-424 M – kDa 425-496 C – – Inputs (2%) IP: GFP C GFP-CEP63 truncation proteins D GFP-CEP63 truncation proteins Inputs (5%) IP: GFP Inputs (5%) IP: GFP kDa kDa 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl Myc- 150- Myc- 100- CCDC14 KIAA0753 100- 100- 75- 75- GFP- GFP- 50- CEP63 50- CEP63 37- 37- Figure S4 A siCtl -

The P53/P73 - P21cip1 Tumor Suppressor Axis Guards Against Chromosomal Instability by Restraining CDK1 in Human Cancer Cells
Oncogene (2021) 40:436–451 https://doi.org/10.1038/s41388-020-01524-4 ARTICLE The p53/p73 - p21CIP1 tumor suppressor axis guards against chromosomal instability by restraining CDK1 in human cancer cells 1 1 2 1 2 Ann-Kathrin Schmidt ● Karoline Pudelko ● Jan-Eric Boekenkamp ● Katharina Berger ● Maik Kschischo ● Holger Bastians 1 Received: 2 July 2020 / Revised: 2 October 2020 / Accepted: 13 October 2020 / Published online: 9 November 2020 © The Author(s) 2020. This article is published with open access Abstract Whole chromosome instability (W-CIN) is a hallmark of human cancer and contributes to the evolvement of aneuploidy. W-CIN can be induced by abnormally increased microtubule plus end assembly rates during mitosis leading to the generation of lagging chromosomes during anaphase as a major form of mitotic errors in human cancer cells. Here, we show that loss of the tumor suppressor genes TP53 and TP73 can trigger increased mitotic microtubule assembly rates, lagging chromosomes, and W-CIN. CDKN1A, encoding for the CDK inhibitor p21CIP1, represents a critical target gene of p53/p73. Loss of p21CIP1 unleashes CDK1 activity which causes W-CIN in otherwise chromosomally stable cancer cells. fi Vice versa 1234567890();,: 1234567890();,: Consequently, induction of CDK1 is suf cient to induce abnormal microtubule assembly rates and W-CIN. , partial inhibition of CDK1 activity in chromosomally unstable cancer cells corrects abnormal microtubule behavior and suppresses W-CIN. Thus, our study shows that the p53/p73 - p21CIP1 tumor suppressor axis, whose loss is associated with W-CIN in human cancer, safeguards against chromosome missegregation and aneuploidy by preventing abnormally increased CDK1 activity. -

Cep57, a NEDD1-Binding Pericentriolar Material Component, Is Essential for Spindle Pole Integrity
Cell Research (2012) :1-12. © 2012 IBCB, SIBS, CAS All rights reserved 1001-0602/12 $ 32.00 npg ORIGINAL ARTICLE www.nature.com/cr Cep57, a NEDD1-binding pericentriolar material component, is essential for spindle pole integrity Qixi Wu1, *, Runsheng He1, *, Haining Zhou1, Albert CH Yu2, Bo Zhang1, Junlin Teng1, Jianguo Chen1, 3 1The State Key Laboratory of Biomembrane and Membrane Bioengineering and The Key Laboratory of Cell Proliferation and Dif- ferentiation of Ministry of Education, College of Life Sciences, Peking University, Beijing 100871, China; 2Department of Neurobi- ology, Neuroscience Research Institute, School of Basic Medical Sciences, Peking University, Beijing 100191, China; 3The Center for Theoretical Biology, Peking University, Beijing 100871, China Formation of a bipolar spindle is indispensable for faithful chromosome segregation and cell division. Spindle in- tegrity is largely dependent on the centrosome and the microtubule network. Centrosome protein Cep57 can bundle microtubules in mammalian cells. Its related protein (Cep57R) in Xenopus was characterized as a stabilization factor for microtubule-kinetochore attachment. Here we show that Cep57 is a pericentriolar material (PCM) component. Its interaction with NEDD1 is necessary for the centrosome localization of Cep57. Depletion of Cep57 leads to unaligned chromosomes and a multipolar spindle, which is induced by PCM fragmentation. In the absence of Cep57, cen- trosome microtubule array assembly activity is weakened, and the spindle length and microtubule density decrease. As a spindle microtubule-binding protein, Cep57 is also responsible for the proper organization of the spindle micro- tubule and localization of spindle pole focusing proteins. Collectively, these results suggest that Cep57, as a NEDD1- binding centrosome component, could function as a spindle pole- and microtubule-stabilizing factor for establishing robust spindle architecture. -

Methods in and Applications of the Sequencing of Short Non-Coding Rnas" (2013)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2013 Methods in and Applications of the Sequencing of Short Non- Coding RNAs Paul Ryvkin University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Genetics Commons, and the Molecular Biology Commons Recommended Citation Ryvkin, Paul, "Methods in and Applications of the Sequencing of Short Non-Coding RNAs" (2013). Publicly Accessible Penn Dissertations. 922. https://repository.upenn.edu/edissertations/922 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/922 For more information, please contact [email protected]. Methods in and Applications of the Sequencing of Short Non-Coding RNAs Abstract Short non-coding RNAs are important for all domains of life. With the advent of modern molecular biology their applicability to medicine has become apparent in settings ranging from diagonistic biomarkers to therapeutics and fields angingr from oncology to neurology. In addition, a critical, recent technological development is high-throughput sequencing of nucleic acids. The convergence of modern biotechnology with developments in RNA biology presents opportunities in both basic research and medical settings. Here I present two novel methods for leveraging high-throughput sequencing in the study of short non- coding RNAs, as well as a study in which they are applied to Alzheimer's Disease (AD). The computational methods presented here include High-throughput Annotation of Modified Ribonucleotides (HAMR), which enables researchers to detect post-transcriptional covalent modifications ot RNAs in a high-throughput manner. In addition, I describe Classification of RNAs by Analysis of Length (CoRAL), a computational method that allows researchers to characterize the pathways responsible for short non-coding RNA biogenesis. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

The Centriole Protein CEP76 Negatively Regulates PLK1 Activity in the Cytoplasm for Proper Mitotic Progression
© 2020. Published by The Company of Biologists Ltd | Journal of Cell Science (2020) 133, jcs241281. doi:10.1242/jcs.241281 RESEARCH ARTICLE The centriole protein CEP76 negatively regulates PLK1 activity in the cytoplasm for proper mitotic progression Yutaka Takeda, Kaho Yamazaki, Kaho Hashimoto, Koki Watanabe, Takumi Chinen*,‡ and Daiju Kitagawa*,‡ ABSTRACT are important for accurate mitotic progression (Golsteyn, 1995; Polo-like kinase 1 (PLK1) dynamically changes its localization and Schmucker and Sumara, 2014). Several factors regulate PLK1 plays important roles in proper mitotic progression. In particular, strict localization during mitosis. PLK1 is recruited to centrosomes in a control of cytoplasmic PLK1 is needed to prevent mitotic defects. CDK1-dependent manner (Lee et al., 2014). The initial recruitment However, the regulation of cytoplasmic PLK1 is not fully understood. In of PLK1 to kinetochores is mediated by its interaction with PBIP1 this study, we show that CEP76, a centriolar protein, physically (also known as CENPU; Kang et al., 2006). During anaphase, PLK1 interacts with PLK1 and tightly controls the activation of cytoplasmic binds to PRC1 and changes its localization to the midbody (Hu et al., PLK1 during mitosis in human cells. We found that removal of 2012). In addition, strict control of PLK1 in the cytoplasm is needed centrosomes induced ectopic aggregation of PLK1, which is highly to prevent its aggregation and mitotic defects (Mukhopadhyay and phosphorylated, in the cytoplasm during mitosis. Importantly, a Dasso, 2010; Schmit et al., 2012; Zhao et al., 2016). However, the targeted RNAi screen revealed that depletion of CEP76 resulted in a detailed mechanisms through which the localization, expression and similar phenotype. -

Is Cep70, a Centrosomal Protein with New Roles in Breast Cancer Dissemination and Metastasis, a Facilitator of Epithelial-Mesenchymal Transition?
Is Cep70, a centrosomal protein with new roles in breast cancer dissemination and metastasis, a facilitator of epithelial-mesenchymal transition? Pedro A. Lazo 1,2 1 Experimental Therapeutics and Translational Oncology Program, Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Científicas (CSIC), Universidad de Salamanca, Salamanca, Spain 2 Instituto de Investigación Biomédica de Salamanca (IBSAL), Hospital Universitario de Salamanca, Salamanca, Spain Running title: Cep70 and EMT Disclosures: None declared. Contact address: [email protected] 1 Introduction Microtubules are driving mechanisms of chromosomes, intracellular organelle movement, and cell shape and motility. Microtubules are organized on centrosomes, which are assembled in microtubule-organizing centers (MTOC). In mitosis there is no nuclear envelope and centromeres associated to microtubules are mainly involved in chromosome redistribution into daughter cells. In differentiated cells the microtubule- organizing centers are dispersed in the cytoplasm (non centrosomal (ncMTOC)) and interact with the minus end of microtubules through γ-tubulin. 1 However, it is not known if the microtubule contribution to tumor biology is only by facilitating tumor aneuploidy. During tumor dissemination, a process not linked to cell division, important changes take place in cell shape and motility. Microtubules are very dynamic because of their inherent structural instability, and this plasticity facilitates their reorganization during the epithelial-mesenchymal -

Integrated Analysis of the Critical Region 5P15.3–P15.2 Associated with Cri-Du-Chat Syndrome
Genetics and Molecular Biology, 42, 1(suppl), 186-196 (2019) Copyright © 2019, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-GMB-2018-0173 Research Article Integrated analysis of the critical region 5p15.3–p15.2 associated with cri-du-chat syndrome Thiago Corrêa1, Bruno César Feltes2 and Mariluce Riegel1,3* 1Post-Graduate Program in Genetics and Molecular Biology, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. 2Institute of Informatics, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. 3Medical Genetics Service, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brazil. Abstract Cri-du-chat syndrome (CdCs) is one of the most common contiguous gene syndromes, with an incidence of 1:15,000 to 1:50,000 live births. To better understand the etiology of CdCs at the molecular level, we investigated theprotein–protein interaction (PPI) network within the critical chromosomal region 5p15.3–p15.2 associated with CdCs using systemsbiology. Data were extracted from cytogenomic findings from patients with CdCs. Based on clin- ical findings, molecular characterization of chromosomal rearrangements, and systems biology data, we explored possible genotype–phenotype correlations involving biological processes connected with CdCs candidate genes. We identified biological processes involving genes previously found to be associated with CdCs, such as TERT, SLC6A3, and CTDNND2, as well as novel candidate proteins with potential contributions to CdCs phenotypes, in- cluding CCT5, TPPP, MED10, ADCY2, MTRR, CEP72, NDUFS6, and MRPL36. Although further functional analy- ses of these proteins are required, we identified candidate proteins for the development of new multi-target genetic editing tools to study CdCs. -

Microtubule Regulation in Cystic Fibrosis Pathophysiology
MICROTUBULE REGULATION IN CYSTIC FIBROSIS PATHOPHYSIOLOGY By: SHARON MARIE RYMUT Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Dr. Thomas J Kelley Department of Pharmacology CASE WESTERN RESERVE UNIVERSITY August 2015 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/ dissertation of Sharon Marie Rymut candidate for the Doctor of Philosophy degree* Dissertation Advisor: Thomas J Kelley Committee Chair: Paul N MacDonald Committee Member: Ruth E Siegel Committee Member: Craig A Hodges Committee Member: Danny Manor Committee Member: Rebecca J Darrah Date of Defense: April 29, 2015 * We also certify that written approval has been obtained for any proprietary material contained therein. ii Dedication There are five chapters in this dissertation. To Mom, Dad, Joe, Marie and Susan, I dedicate one chapter to each of you. You can fight about which chapter you want later. iii Table of Contents Table of Contents................................................................................................................iv List of Tables .................................................................................................................... vii List of Figures .................................................................................................................. viii Acknowledgements ............................................................................................................. x List of Abbreviations ....................................................................................................... -
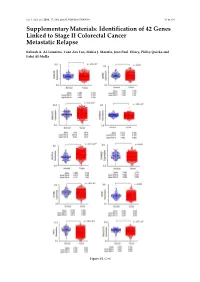
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

The Transformation of the Centrosome Into the Basal Body: Similarities and Dissimilarities Between Somatic and Male Germ Cells and Their Relevance for Male Fertility
cells Review The Transformation of the Centrosome into the Basal Body: Similarities and Dissimilarities between Somatic and Male Germ Cells and Their Relevance for Male Fertility Constanza Tapia Contreras and Sigrid Hoyer-Fender * Göttingen Center of Molecular Biosciences, Johann-Friedrich-Blumenbach Institute for Zoology and Anthropology-Developmental Biology, Faculty of Biology and Psychology, Georg-August University of Göttingen, 37077 Göttingen, Germany; [email protected] * Correspondence: [email protected] Abstract: The sperm flagellum is essential for the transport of the genetic material toward the oocyte and thus the transmission of the genetic information to the next generation. During the haploid phase of spermatogenesis, i.e., spermiogenesis, a morphological and molecular restructuring of the male germ cell, the round spermatid, takes place that includes the silencing and compaction of the nucleus, the formation of the acrosomal vesicle from the Golgi apparatus, the formation of the sperm tail, and, finally, the shedding of excessive cytoplasm. Sperm tail formation starts in the round spermatid stage when the pair of centrioles moves toward the posterior pole of the nucleus. The sperm tail, eventually, becomes located opposed to the acrosomal vesicle, which develops at the anterior pole of the nucleus. The centriole pair tightly attaches to the nucleus, forming a nuclear membrane indentation. An Citation: Tapia Contreras, C.; articular structure is formed around the centriole pair known as the connecting piece, situated in the Hoyer-Fender, S. The Transformation neck region and linking the sperm head to the tail, also named the head-to-tail coupling apparatus or, of the Centrosome into the Basal in short, HTCA.