Statistical Mechanics 215A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Key Concepts for Future QIS Learners Workshop Output Published Online May 13, 2020
Key Concepts for Future QIS Learners Workshop output published online May 13, 2020 Background and Overview On behalf of the Interagency Working Group on Workforce, Industry and Infrastructure, under the NSTC Subcommittee on Quantum Information Science (QIS), the National Science Foundation invited 25 researchers and educators to come together to deliberate on defining a core set of key concepts for future QIS learners that could provide a starting point for further curricular and educator development activities. The deliberative group included university and industry researchers, secondary school and college educators, and representatives from educational and professional organizations. The workshop participants focused on identifying concepts that could, with additional supporting resources, help prepare secondary school students to engage with QIS and provide possible pathways for broader public engagement. This workshop report identifies a set of nine Key Concepts. Each Concept is introduced with a concise overall statement, followed by a few important fundamentals. Connections to current and future technologies are included, providing relevance and context. The first Key Concept defines the field as a whole. Concepts 2-6 introduce ideas that are necessary for building an understanding of quantum information science and its applications. Concepts 7-9 provide short explanations of critical areas of study within QIS: quantum computing, quantum communication and quantum sensing. The Key Concepts are not intended to be an introductory guide to quantum information science, but rather provide a framework for future expansion and adaptation for students at different levels in computer science, mathematics, physics, and chemistry courses. As such, it is expected that educators and other community stakeholders may not yet have a working knowledge of content covered in the Key Concepts. -

Canonical Ensemble
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 8. Canonical Ensemble January 26, 2011 Contents Outline • In this chapter, we will establish the equilibrium statistical distribution for systems maintained at a constant temperature T , through thermal contact with a heat bath. • The resulting distribution is also called Boltzmann's distribution. • The canonical distribution also leads to definition of the partition function and an expression for Helmholtz free energy, analogous to Boltzmann's Entropy formula. • We will study energy fluctuation at constant temperature, and witness another fluctuation- dissipation theorem (FDT) and finally establish the equivalence of micro canonical ensemble and canonical ensemble in the thermodynamic limit. (We first met a mani- festation of FDT in diffusion as Einstein's relation.) Reading Assignment: Reif x6.1-6.7, x6.10 1 1 Temperature For an isolated system, with fixed N { number of particles, V { volume, E { total energy, it is most conveniently described by the microcanonical (NVE) ensemble, which is a uniform distribution between two constant energy surfaces. const E ≤ H(fq g; fp g) ≤ E + ∆E ρ (fq g; fp g) = i i (1) mc i i 0 otherwise Statistical mechanics also provides the expression for entropy S(N; V; E) = kB ln Ω. In thermodynamics, S(N; V; E) can be transformed to a more convenient form (by Legendre transform) of Helmholtz free energy A(N; V; T ), which correspond to a system with constant N; V and temperature T . Q: Does the transformation from N; V; E to N; V; T have a meaning in statistical mechanics? A: The ensemble of systems all at constant N; V; T is called the canonical NVT ensemble. -

Identical Particles
8.06 Spring 2016 Lecture Notes 4. Identical particles Aram Harrow Last updated: May 19, 2016 Contents 1 Fermions and Bosons 1 1.1 Introduction and two-particle systems .......................... 1 1.2 N particles ......................................... 3 1.3 Non-interacting particles .................................. 5 1.4 Non-zero temperature ................................... 7 1.5 Composite particles .................................... 7 1.6 Emergence of distinguishability .............................. 9 2 Degenerate Fermi gas 10 2.1 Electrons in a box ..................................... 10 2.2 White dwarves ....................................... 12 2.3 Electrons in a periodic potential ............................. 16 3 Charged particles in a magnetic field 21 3.1 The Pauli Hamiltonian ................................... 21 3.2 Landau levels ........................................ 23 3.3 The de Haas-van Alphen effect .............................. 24 3.4 Integer Quantum Hall Effect ............................... 27 3.5 Aharonov-Bohm Effect ................................... 33 1 Fermions and Bosons 1.1 Introduction and two-particle systems Previously we have discussed multiple-particle systems using the tensor-product formalism (cf. Section 1.2 of Chapter 3 of these notes). But this applies only to distinguishable particles. In reality, all known particles are indistinguishable. In the coming lectures, we will explore the mathematical and physical consequences of this. First, consider classical many-particle systems. If a single particle has state described by position and momentum (~r; p~), then the state of N distinguishable particles can be written as (~r1; p~1; ~r2; p~2;:::; ~rN ; p~N ). The notation (·; ·;:::; ·) denotes an ordered list, in which different posi tions have different meanings; e.g. in general (~r1; p~1; ~r2; p~2)6 = (~r2; p~2; ~r1; p~1). 1 To describe indistinguishable particles, we can use set notation. -
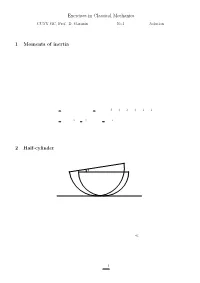
Exercises in Classical Mechanics 1 Moments of Inertia 2 Half-Cylinder
Exercises in Classical Mechanics CUNY GC, Prof. D. Garanin No.1 Solution ||||||||||||||||||||||||||||||||||||||||| 1 Moments of inertia (5 points) Calculate tensors of inertia with respect to the principal axes of the following bodies: a) Hollow sphere of mass M and radius R: b) Cone of the height h and radius of the base R; both with respect to the apex and to the center of mass. c) Body of a box shape with sides a; b; and c: Solution: a) By symmetry for the sphere I®¯ = I±®¯: (1) One can ¯nd I easily noticing that for the hollow sphere is 1 1 X ³ ´ I = (I + I + I ) = m y2 + z2 + z2 + x2 + x2 + y2 3 xx yy zz 3 i i i i i i i i 2 X 2 X 2 = m r2 = R2 m = MR2: (2) 3 i i 3 i 3 i i b) and c): Standard solutions 2 Half-cylinder ϕ (10 points) Consider a half-cylinder of mass M and radius R on a horizontal plane. a) Find the position of its center of mass (CM) and the moment of inertia with respect to CM. b) Write down the Lagrange function in terms of the angle ' (see Fig.) c) Find the frequency of cylinder's oscillations in the linear regime, ' ¿ 1. Solution: (a) First we ¯nd the distance a between the CM and the geometrical center of the cylinder. With σ being the density for the cross-sectional surface, so that ¼R2 M = σ ; (3) 2 1 one obtains Z Z 2 2σ R p σ R q a = dx x R2 ¡ x2 = dy R2 ¡ y M 0 M 0 ¯ 2 ³ ´3=2¯R σ 2 2 ¯ σ 2 3 4 = ¡ R ¡ y ¯ = R = R: (4) M 3 0 M 3 3¼ The moment of inertia of the half-cylinder with respect to the geometrical center is the same as that of the cylinder, 1 I0 = MR2: (5) 2 The moment of inertia with respect to the CM I can be found from the relation I0 = I + Ma2 (6) that yields " µ ¶ # " # 1 4 2 1 32 I = I0 ¡ Ma2 = MR2 ¡ = MR2 1 ¡ ' 0:3199MR2: (7) 2 3¼ 2 (3¼)2 (b) The Lagrange function L is given by L('; '_ ) = T ('; '_ ) ¡ U('): (8) The potential energy U of the half-cylinder is due to the elevation of its CM resulting from the deviation of ' from zero. -

A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Article A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action Ivan Kennedy 1,2,*, Harold Geering 2, Michael Rose 3 and Angus Crossan 2 1 Sydney Institute of Agriculture, University of Sydney, NSW 2006, Australia 2 QuickTest Technologies, PO Box 6285 North Ryde, NSW 2113, Australia; [email protected] (H.G.); [email protected] (A.C.) 3 NSW Department of Primary Industries, Wollongbar NSW 2447, Australia; [email protected] * Correspondence: [email protected]; Tel.: + 61-4-0794-9622 Received: 23 March 2019; Accepted: 26 April 2019; Published: 1 May 2019 Abstract: A convenient practical model for accurately estimating the total entropy (ΣSi) of atmospheric gases based on physical action is proposed. This realistic approach is fully consistent with statistical mechanics, but reinterprets its partition functions as measures of translational, rotational, and vibrational action or quantum states, to estimate the entropy. With all kinds of molecular action expressed as logarithmic functions, the total heat required for warming a chemical system from 0 K (ΣSiT) to a given temperature and pressure can be computed, yielding results identical with published experimental third law values of entropy. All thermodynamic properties of gases including entropy, enthalpy, Gibbs energy, and Helmholtz energy are directly estimated using simple algorithms based on simple molecular and physical properties, without resource to tables of standard values; both free energies are measures of quantum field states and of minimal statistical degeneracy, decreasing with temperature and declining density. We propose that this more realistic approach has heuristic value for thermodynamic computation of atmospheric profiles, based on steady state heat flows equilibrating with gravity. -
![Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals](https://docslib.b-cdn.net/cover/4942/arxiv-1910-10745v1-cond-mat-str-el-23-oct-2019-2-2-symmetry-protected-time-crystals-304942.webp)
Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals
A Brief History of Time Crystals Vedika Khemania,b,∗, Roderich Moessnerc, S. L. Sondhid aDepartment of Physics, Harvard University, Cambridge, Massachusetts 02138, USA bDepartment of Physics, Stanford University, Stanford, California 94305, USA cMax-Planck-Institut f¨urPhysik komplexer Systeme, 01187 Dresden, Germany dDepartment of Physics, Princeton University, Princeton, New Jersey 08544, USA Abstract The idea of breaking time-translation symmetry has fascinated humanity at least since ancient proposals of the per- petuum mobile. Unlike the breaking of other symmetries, such as spatial translation in a crystal or spin rotation in a magnet, time translation symmetry breaking (TTSB) has been tantalisingly elusive. We review this history up to recent developments which have shown that discrete TTSB does takes place in periodically driven (Floquet) systems in the presence of many-body localization (MBL). Such Floquet time-crystals represent a new paradigm in quantum statistical mechanics — that of an intrinsically out-of-equilibrium many-body phase of matter with no equilibrium counterpart. We include a compendium of the necessary background on the statistical mechanics of phase structure in many- body systems, before specializing to a detailed discussion of the nature, and diagnostics, of TTSB. In particular, we provide precise definitions that formalize the notion of a time-crystal as a stable, macroscopic, conservative clock — explaining both the need for a many-body system in the infinite volume limit, and for a lack of net energy absorption or dissipation. Our discussion emphasizes that TTSB in a time-crystal is accompanied by the breaking of a spatial symmetry — so that time-crystals exhibit a novel form of spatiotemporal order. -

LECTURES on MATHEMATICAL STATISTICAL MECHANICS S. Adams
ISSN 0070-7414 Sgr´ıbhinn´ıInstiti´uid Ard-L´einnBhaile´ Atha´ Cliath Sraith. A. Uimh 30 Communications of the Dublin Institute for Advanced Studies Series A (Theoretical Physics), No. 30 LECTURES ON MATHEMATICAL STATISTICAL MECHANICS By S. Adams DUBLIN Institi´uid Ard-L´einnBhaile´ Atha´ Cliath Dublin Institute for Advanced Studies 2006 Contents 1 Introduction 1 2 Ergodic theory 2 2.1 Microscopic dynamics and time averages . .2 2.2 Boltzmann's heuristics and ergodic hypothesis . .8 2.3 Formal Response: Birkhoff and von Neumann ergodic theories9 2.4 Microcanonical measure . 13 3 Entropy 16 3.1 Probabilistic view on Boltzmann's entropy . 16 3.2 Shannon's entropy . 17 4 The Gibbs ensembles 20 4.1 The canonical Gibbs ensemble . 20 4.2 The Gibbs paradox . 26 4.3 The grandcanonical ensemble . 27 4.4 The "orthodicity problem" . 31 5 The Thermodynamic limit 33 5.1 Definition . 33 5.2 Thermodynamic function: Free energy . 37 5.3 Equivalence of ensembles . 42 6 Gibbs measures 44 6.1 Definition . 44 6.2 The one-dimensional Ising model . 47 6.3 Symmetry and symmetry breaking . 51 6.4 The Ising ferromagnet in two dimensions . 52 6.5 Extreme Gibbs measures . 57 6.6 Uniqueness . 58 6.7 Ergodicity . 60 7 A variational characterisation of Gibbs measures 62 8 Large deviations theory 68 8.1 Motivation . 68 8.2 Definition . 70 8.3 Some results for Gibbs measures . 72 i 9 Models 73 9.1 Lattice Gases . 74 9.2 Magnetic Models . 75 9.3 Curie-Weiss model . 77 9.4 Continuous Ising model . -

Entropy and H Theorem the Mathematical Legacy of Ludwig Boltzmann
ENTROPY AND H THEOREM THE MATHEMATICAL LEGACY OF LUDWIG BOLTZMANN Cambridge/Newton Institute, 15 November 2010 C´edric Villani University of Lyon & Institut Henri Poincar´e(Paris) Cutting-edge physics at the end of nineteenth century Long-time behavior of a (dilute) classical gas Take many (say 1020) small hard balls, bouncing against each other, in a box Let the gas evolve according to Newton’s equations Prediction by Maxwell and Boltzmann The distribution function is asymptotically Gaussian v 2 f(t, x, v) a exp | | as t ≃ − 2T → ∞ Based on four major conceptual advances 1865-1875 Major modelling advance: Boltzmann equation • Major mathematical advance: the statistical entropy • Major physical advance: macroscopic irreversibility • Major PDE advance: qualitative functional study • Let us review these advances = journey around centennial scientific problems ⇒ The Boltzmann equation Models rarefied gases (Maxwell 1865, Boltzmann 1872) f(t, x, v) : density of particles in (x, v) space at time t f(t, x, v) dxdv = fraction of mass in dxdv The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution -

Bose-Einstein Condensation of Photons and Grand-Canonical Condensate fluctuations
Bose-Einstein condensation of photons and grand-canonical condensate fluctuations Jan Klaers Institute for Applied Physics, University of Bonn, Germany Present address: Institute for Quantum Electronics, ETH Zürich, Switzerland Martin Weitz Institute for Applied Physics, University of Bonn, Germany Abstract We review recent experiments on the Bose-Einstein condensation of photons in a dye-filled optical microresonator. The most well-known example of a photon gas, pho- tons in blackbody radiation, does not show Bose-Einstein condensation. Instead of massively populating the cavity ground mode, photons vanish in the cavity walls when they are cooled down. The situation is different in an ultrashort optical cavity im- printing a low-frequency cutoff on the photon energy spectrum that is well above the thermal energy. The latter allows for a thermalization process in which both tempera- ture and photon number can be tuned independently of each other or, correspondingly, for a non-vanishing photon chemical potential. We here describe experiments demon- strating the fluorescence-induced thermalization and Bose-Einstein condensation of a two-dimensional photon gas in the dye microcavity. Moreover, recent measurements on the photon statistics of the condensate, showing Bose-Einstein condensation in the grandcanonical ensemble limit, will be reviewed. 1 Introduction Quantum statistical effects become relevant when a gas of particles is cooled, or its den- sity is increased, to the point where the associated de Broglie wavepackets spatially over- arXiv:1611.10286v1 [cond-mat.quant-gas] 30 Nov 2016 lap. For particles with integer spin (bosons), the phenomenon of Bose-Einstein condensation (BEC) then leads to macroscopic occupation of a single quantum state at finite tempera- tures [1]. -

Magnetic Materials I
5 Magnetic materials I Magnetic materials I ● Diamagnetism ● Paramagnetism Diamagnetism - susceptibility All materials can be classified in terms of their magnetic behavior falling into one of several categories depending on their bulk magnetic susceptibility χ. without spin M⃗ 1 χ= in general the susceptibility is a position dependent tensor M⃗ (⃗r )= ⃗r ×J⃗ (⃗r ) H⃗ 2 ] In some materials the magnetization is m / not a linear function of field strength. In A [ such cases the differential susceptibility M is introduced: d M⃗ χ = d d H⃗ We usually talk about isothermal χ susceptibility: ∂ M⃗ χ = T ( ∂ ⃗ ) H T Theoreticians define magnetization as: ∂ F⃗ H[A/m] M=− F=U−TS - Helmholtz free energy [4] ( ∂ H⃗ ) T N dU =T dS− p dV +∑ μi dni i=1 N N dF =T dS − p dV +∑ μi dni−T dS−S dT =−S dT − p dV +∑ μi dni i=1 i=1 Diamagnetism - susceptibility It is customary to define susceptibility in relation to volume, mass or mole: M⃗ (M⃗ /ρ) m3 (M⃗ /mol) m3 χ= [dimensionless] , χρ= , χ = H⃗ H⃗ [ kg ] mol H⃗ [ mol ] 1emu=1×10−3 A⋅m2 The general classification of materials according to their magnetic properties μ<1 <0 diamagnetic* χ ⃗ ⃗ ⃗ ⃗ ⃗ ⃗ B=μ H =μr μ0 H B=μo (H +M ) μ>1 >0 paramagnetic** ⃗ ⃗ ⃗ χ μr μ0 H =μo( H + M ) → μ r=1+χ μ≫1 χ≫1 ferromagnetic*** *dia /daɪə mæ ɡˈ n ɛ t ɪ k/ -Greek: “from, through, across” - repelled by magnets. We have from L2: 1 2 the force is directed antiparallel to the gradient of B2 F = V ∇ B i.e. -

Statistical Physics– a Second Course
Statistical Physics– a second course Finn Ravndal and Eirik Grude Flekkøy Department of Physics University of Oslo September 3, 2008 2 Contents 1 Summary of Thermodynamics 5 1.1 Equationsofstate .......................... 5 1.2 Lawsofthermodynamics. 7 1.3 Maxwell relations and thermodynamic derivatives . .... 9 1.4 Specificheatsandcompressibilities . 10 1.5 Thermodynamicpotentials . 12 1.6 Fluctuations and thermodynamic stability . .. 15 1.7 Phasetransitions ........................... 16 1.8 EntropyandGibbsParadox. 18 2 Non-Interacting Particles 23 1 2.1 Spin- 2 particlesinamagneticfield . 23 2.2 Maxwell-Boltzmannstatistics . 28 2.3 Idealgas................................ 32 2.4 Fermi-Diracstatistics. 35 2.5 Bose-Einsteinstatistics. 36 3 Statistical Ensembles 39 3.1 Ensemblesinphasespace . 39 3.2 Liouville’stheorem . .. .. .. .. .. .. .. .. .. .. 42 3.3 Microcanonicalensembles . 45 3.4 Free particles and multi-dimensional spheres . .... 48 3.5 Canonicalensembles . 50 3.6 Grandcanonicalensembles . 54 3.7 Isobaricensembles .......................... 58 3.8 Informationtheory . .. .. .. .. .. .. .. .. .. .. 62 4 Real Gases and Liquids 67 4.1 Correlationfunctions. 67 4.2 Thevirialtheorem .......................... 73 4.3 Mean field theory for the van der Waals equation . 76 4.4 Osmosis ................................ 80 3 4 CONTENTS 5 Quantum Gases and Liquids 83 5.1 Statisticsofidenticalparticles. .. 83 5.2 Blackbodyradiationandthephotongas . 88 5.3 Phonons and the Debye theory of specific heats . 96 5.4 Bosonsatnon-zerochemicalpotential . -

Grand-Canonical Ensemble
PHYS4006: Thermal and Statistical Physics Lecture Notes (Unit - IV) Open System: Grand-canonical Ensemble Dr. Neelabh Srivastava (Assistant Professor) Department of Physics Programme: M.Sc. Physics Mahatma Gandhi Central University Semester: 2nd Motihari-845401, Bihar E-mail: [email protected] • In microcanonical ensemble, each system contains same fixed energy as well as same number of particles. Hence, the system dealt within this ensemble is a closed isolated system. • With microcanonical ensemble, we can not deal with the systems that are kept in contact with a heat reservoir at a given temperature. 2 • In canonical ensemble, the condition of constant energy is relaxed and the system is allowed to exchange energy but not the particles with the system, i.e. those systems which are not isolated but are in contact with a heat reservoir. • This model could not be applied to those processes in which number of particle varies, i.e. chemical process, nuclear reactions (where particles are created and destroyed) and quantum process. 3 • So, for the method of ensemble to be applicable to such processes where number of particles as well as energy of the system changes, it is necessary to relax the condition of fixed number of particles. 4 • Such an ensemble where both the energy as well as number of particles can be exchanged with the heat reservoir is called Grand Canonical Ensemble. • In canonical ensemble, T, V and N are independent variables. Whereas, in grand canonical ensemble, the system is described by its temperature (T),volume (V) and chemical potential (μ). 5 • Since, the system is not isolated, its microstates are not equally probable.