Fe6+) AS an ALTERNATIVE METHOD FOR
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446
Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, D.C. 20460 OFFICE OF CHEMICAL SAFETY AND POLLUTION PREVENTION MEMORANDUM Date: March 11, 2020 SUBJECT: Registration Review Draft Risk Assessment for the Peroxy Compounds PC Code: 000595, 063201, 063604, 063607, DP Barcode: 455445, 455446 063209, 128860 Decision No: 558073, 558074 Docket No: EPA-HQ-OPP-2009-0546 Regulatory Action: Registration Review Case No: 6059, 4072, 5081 Risk Assessment Type: DRA CAS No: 7722-84-1, 79-21-0, 33734-57-5, 15630-89-4, 10058-23-8, 70693-62-8 TO: Kendall Ziner, Chemical Review Manager Rick Fehir, Ph.D., Team Lead Rose Kyprianou, Branch Chief Regulatory Management Branch (RMB) II Antimicrobials Division (7510P) Office of Pesticide Programs FROM: Andrew Byro, Ph.D., Chemist Kathryn Korthauer, Biologist Timothy Dole, Industrial Hygienist Deborah Burgin, Ph.D., DABT, Toxicologist Risk Assessment and Science Support Branch Antimicrobials Division (7510P) Office of Pesticide Programs THROUGH: Judy Facey, Ph.D., Human Health Risk Assessment Process Leader MP for JF Diana Hsieh, Ecological Risk Assessment Process Leader MP for DH Timothy Leighton, Senior Science Advisor MP for TL Laura Parsons, Associate Branch Chief Melissa Panger, Ph.D., Branch Chief Risk Assessment and Science Support Branch Antimicrobials Division (7510P) This document provides the draft human health and ecological risk assessment conducted in support of the antimicrobial use sites of the following peroxy compounds: hydrogen peroxide, peracetic acid, peroxyoctanoic acid, and sodium percarbonate. Page 1 of 74 Peroxy Compounds Human Health and Ecological Draft Risk Assessment DP 455445, 455446 Although the peroxymonosulfate compounds were included in the peroxy compounds Final Work Plan (FWP), they will not be included in this risk assessment. -

Decolorization of Reactive Orange 16 Via Ferrate(VI) Oxidation Assisted by Sonication
Turkish Journal of Chemistry Turk J Chem (2017) 41: 577 { 586 http://journals.tubitak.gov.tr/chem/ ⃝c TUB¨ ITAK_ Research Article doi:10.3906/kim-1701-8 Decolorization of reactive orange 16 via ferrate(VI) oxidation assisted by sonication Serkan S¸AHINKAYA_ ∗ Department of Environmental Engineering, Faculty of Engineering and Architecture, Nev¸sehirHacı Bekta¸sVeli University, Nev¸sehir,Turkey Received: 02.01.2017 • Accepted/Published Online: 24.03.2017 • Final Version: 05.09.2017 Abstract: The decolorization of azo dye C.I. reactive orange 16 (RO 16) via ferrate(VI) and sono-ferrate(VI) methods, which is the combination of the ferrate(VI) oxidation method with sonication, has been achieved in the present study. The influences of some important operating parameters, which are the initial pH, the concentration of potassium ferrate(VI) (K 2 FeO 4) and the RO 16 dye, and ultrasonic density (for only the sono-ferrate(VI) method), on the color removal have −1 been investigated. The optimum conditions have been determined as pH = 7 and [K 2 FeO 4 ] = 50 mg L for the −1 −1 individual ferrate(VI) oxidation method and pH = 7 and [K 2 FeO 4 ] = 50 mg L by direct sonication at 0.50 W mL ultrasonic density and 20 kHz fixed frequency for the sono-ferrate(VI) method. The color removal efficiencies were 85% by ferrate(VI) method and 91% by sono-ferrate(VI) method. Kinetic studies were also performed for the decolorization of RO 16 under the optimized conditions at room temperature. It was seen that the oxidative decolorization of RO 16 via the sono-ferrate(VI) method happened more rapidly because of the production of OH • radical through sonication compared to the individual ferrate(VI) method. -

Determination of Tetrathionate, Thiosulfate, Sulfite and Trithionate in Their Mixtures by Spectrophotometry
ANALYTICAL SCIENCES FEBRUARY 1989, VOL. 5 79 Determination of Tetrathionate, Thiosulfate, Sulfite and Trithionate in Their Mixtures by Spectrophotometry Tomozo KOH, Yasuyuki MIURA, Masahiro IsHIMORI and Norihito YAMAMURO Department of Chemistry, Faculty of Science, Tokai University, Hiratsuka, Kanagawa 259-12, Japan The proposed method consists of four procedures; excess iodine for reactions with thiosulfate and/or sulfite under Procedures I, II and III, and the thiocyanate formed under Procedure IV are measured spectrophotometrically after proper chemical treatments. The absorbance obtained by Procedure I corresponds to the sum of the amount of tetrathionate and that of thiosulfate in the mixture. The absorbance obtained by Procedure II corresponds only to the amount of thiosulfate in the mixture. The absorbance obtained by Procedure III corresponds to the sum of the amount of thiosulfate and twice that of sulfite in the mixture. The absorbance obtained by Procedure IV corresponds to the sum of the amount of both thiosulfate and trithionate and twice that of tetrathionate in the mixture. The proposed method was applied to the determination of tetrathionate, thiosulfate, sulfite and trithionate mixed in various ratios in amounts of more than 0.05 µmol with an error below ±0.02 µmol. Keywords Polythionates determination, tetrathionate sulfitolysis, trithionate cyanolysis, tetrathionate-trithionate- thiosulfate-sulfite mixture, spectrophotometry The determination of various sulfur anion species in of four sulfur species of tetrathionate, thiosulfate, their mixtures is desirable for interpreting their redox sulfite and trithionate in a mixture. chemistry in aqueous solution systems. However, this is difficult owing to similarities in their chemical and physical properties. Spectrophotometric methods"2 for Experimental the determination of trithionate, thiosulfate and tetra- thionate in a mixture, based on the formation of Reagents and apparatus thiocyanate by their cyanolysis, have been proposed. -

Life Cycle Assessment of Process Strategies for Value Recovery and Environmental Mitigation of Mining Waste
Life cycle assessment of process strategies for value recovery and environmental mitigation of mining waste by Kieran Hillmar Cairncross Thesis presented in partial fulfilment of the requirements for the Degree of MASTER OF ENGINEERING (CHEMICAL ENGINEERING) in the Faculty of Engineering at Stellenbosch University The financial assistance of the National Research Foundation (NRF) towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at, are those of the author and are not necessarily to be attributed to the NRF. Supervisor Dr Margreth Tadie December 2020 Stellenbosch University https://scholar.sun.ac.za Declaration By submitting this thesis electronically, I declare that the entirety of the work contained therein is my own, original work, that I am the sole author thereof (save to the extent explicitly otherwise stated), that reproduction and publication thereof by Stellenbosch University will not infringe any third party rights and that I have not previously in its entirety or in part submitted it for obtaining any qualification. Date: 11 September 2020 Copyright © 2020 Stellenbosch University All rights reserved i Stellenbosch University https://scholar.sun.ac.za Abstract By 2031, South African primary ore gold grades are forecasted to decline to the gold grades expected in mine tailings resources. Furthermore, the reprocessing of mine tailings does not require the costly excavation and size reduction unit operations necessary for primary ore resources. Mine tailings can therefore be viewed as a secondary gold resource. Hazardous pollutants and acid mine drainage (AMD) emanating from Witwatersrand stockpiled tailings dams affect human and ecosystem health. Potential exists to valorise mine tailings to the circular economy as construction raw materials and mine backfill. -

WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 3/04 (2006.01) A61K 33/40 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/10 (2006.01) A61K 38/44 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 35/74 (2015.01) A61K 31/17 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 16/034973 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 3 1 May 2016 (3 1.05.2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/169,480 1 June 2015 (01 .06.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/327,283 25 April 2016 (25.04.2016) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: XENO BIOSCIENCES INC. -
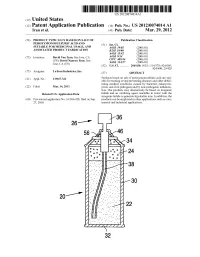
Us 2012/0074.014 A1 2 .. 1
US 2012O074O14A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0074.014 A1 Tran et al. (43) Pub. Date: Mar. 29, 2012 (54) PRODUCT TYPICALLY BASED ON SALT OF Publication Classification PEROXYMONOSULFURIC ACID AND (51) Int. Cl SUITABLE FOR MEDICINAL USAGE, AND iBio/02 (2006.01) ASSOCATED PRODUCT FABRICATION B23P 19/00 (2006.01) A633/42 (2006.01) (75) Inventors: David Van Tran, San Jose, CA A6IR 9/14 (2006.01) (US); David Nguyen Tran, San CD7C 409/244 (2006.01) Jose, CA (US) A6II 3/327 (2006.01) s (52) U.S. Cl. ............. 206/438: 562/1; 514/578; 424/605; 424/400; 29/428 (73) Assignee: LuTran Industries, Inc. (57) ABSTRACT 21) Appl. No.: 13AO47,742 Products based on salt of peroxyperoxVmonosulfuric acid are suit (21) Appl. No 9 able for treating or/and preventing diseases and other debili tating medical conditions caused by bacterial, eukaryotic, (22) Filed: Mar. 14, 2011 prion, and viral pathogens and by non-pathogenic inflamma tion. The products may alternatively be based on inorganic Related U.S. Application Data halide and an oxidizing agent reactable in water with the inorganic halide to generate hypohalite ions. In addition, the (60) Provisional application No. 61/386,928, filed on Sep. products can be employed in other applications such as com 27, 2010. mercial and industrial applications. 5T. 2 .. 2. 1.4. J., Patent Application Publication Mar. 29, 2012 Sheet 1 of 2 US 2012/0074014 A1 cro Q-D 22 Fig. 2a Fig.2b Patent Application Publication Mar. 29, 2012 Sheet 2 of 2 US 2012/0074014 A1 90 92 US 2012/0074014 A1 Mar. -

Book of Abstracts Vol. II
Symposium A Advanced Materials: From Fundamentals to Applications INVITED LECTURES 1. Potassium hydroxide 2. Potassium hydride 3. Potassium carbonate 4. Sodium hydroxide 5. Sodium hydride 6. Sodium carbonate 7. Calcium hydroxide 8. Calcium carbonate 9. Calcium sulphate 10. Calcium nitrate 11. Calcium chloride 12. Barium hydroxide 13. Barium carbonate 14. Barium sulphate 15. Barium nitrate 16. Barium chloride 17. Aluminium sulphate 18. Aluminium nitrate 19. Aluminium chloride 20. Alum 21. Potassium silicate 22. Potassium silicate 23. Potassium calcium silicate 24. Potassium barium silicate 25. Silicon fluoride 26. Ammonium potassium 27. Ethylene chloride compound Chemical symbols used by Dalton, 19th century ← Previous page: Distilling apparatus from John French’s The art of distillation, London 1651 Symposium A: Advanced Materials A - IL 1 Binuclear Complexes as Tectons in Designing Supramolecular Solid-State Architectures Marius Andruh University of Bucharest, Faculty of Chemistry, Inorganic Chemistry Laboratory Str. Dumbrava Rosie nr. 23, 020464-Bucharest, Romania [email protected] We are currently developing a research project on the use of homo- and heterobinuclear complexes as building-blocks in designing both oligonuclear species and high-dimensionality coordination polymers with interesting magnetic properties. The building-blocks are stable binuclear complexes, where the metal ions are held together by compartmental ligands, or alkoxo- bridged copper(II) complexes. The binuclear nodes are connected through appropriate exo- - 3- III bidentate ligands, or through metal-containing anions (e. g. [Cr(NH3)2(NCS)4] , [M(CN)6] , M = Fe , CrIII). A rich variety of 3d-3d and 3d-4f heterometallic complexes, with interesting architectures and topologies of the spin carriers, has been obtained1. -

Energy Efficiency in the MENA Region Good Practices from ACWUA Members
READER Energy Efficiency in the MENA region Good practices from ACWUA members i Reader Energy Efficiency in the MENA region Good practices from ACWUA members Published by The Arab Countries Water Utilities Association (ACWUA) under the guidance of the ACWUA Task Force Energy Efficiency with support from the Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH Registered offices ACWUA in Amman, Jordan GIZ in Bonn and Eschborn, Germany Implemented by Strengthening the MENA water sector through regional networking and training (ACWUA WANT) program www.mena-water.net or www.acwua.org/training or www.acwua.org/node/413 Arab Countries Water Utilities Association (ACWUA) 19A, Umm Umarah Street Al-Rashed Area P.O.Box 962449 Amman 11196 Jordan and Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ) GmbH 65760 Eschborn, Germany Dag-Hammarskjöld-Weg 1-5 Division 3300 Near East On behalf of the German Federal Ministry for Economic Cooperation and Development (BMZ) Editor Hans Hartung, Weikersheim. Germany Program supervision Thomas Petermann (GIZ Germany) Abdellatif Biad (ONEE, Chairperson ACWUA Task Force Energy Efficiency) Mustafa Nasereddin (ACWUA Jordan) Photo credits Thomas Petermann Disclaimer The material in this publication was written by, and reflects the views of the authors. None of the material implies an opinion of any form by GIZ or ACWUA. The information in this book is distributed on an “As is” basis, without warranty. Copyrights September 2015 ACWUA and GIZ ii ENERGY EFFICIENCY READER Energy Efficiency – A strategic objective and assigned target of ACWUA In most cases energy is the number one cost within all water and wastewater service utilities O&M costs, and a controllable one at that. -

“Treatment of Produced Water Using Ferrate (Vi) And
“TREATMENT OF PRODUCED WATER USING FERRATE (VI) AND DIRECTIONAL SOLVENT EXTRACTION.” A Thesis By SEAN X. THIMONS Submitted to Graduate Studies & Office of Research Texas A & M University-San Antonio in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Spring 2021 Major Subject: Water Resources, Science & Technology Water Resources Science and Technology THESIS APPROVAL FORM Student’s Name: Sean X. Thimons (Name must match student records) Date of Defense: (mm/dd/yy or Exempt): 04/21/2021 Today’s Date (mm/dd/yy): 04/21/2021 Anticipated Date of Graduation (Month Year): May, 2021 Major Subject: Water Resources Science and Technology Thesis Title: Treatment of Produced Water Using Ferrate (VI) and Directional Solvent Extraction We the undersigned duly appointed committee have read and examined this manuscript. We certify it is adequate in scope and quality as a thesis for this master's degree and indicate our approval of the content of the document to be submitted to the Office of Research and Graduate Studies for acceptance, or we indicate our dissent below. A vote by all members of the committee with at most one dissension is required to pass. Approve Disapprove Walter Den, Ph.D. Committee Chair Signature Drew Johnson, Ph.D. Approve Disapprove Committee Member Signature Approve Disapprove Virender K. Sharma, Ph.D. VKSharma Committee Member Signature Approve Disapprove Marvin M.F. Lutnesky, Ph.D. Committee Member Signature Approve Disapprove Committee Member Signature Mirley Balasubramanya, Ph.D. Approve Disapprove Department Head Signature Approve Disapprove Vijay Golla, Ph.D. Vice Provost for Research Signature & Graduate Studies Committee Decision: Thesis v Approve Disapprove The student must submit this signed approval form and a PDF file of the thesis to the Office of Research and Graduate Studies. -

Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-Free Bleaching of Hardwoods Prehydrolysis-Kraft Pulps
Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps 著者 RIZALUDDIN Andri Taufick year 2016 その他のタイトル 広葉樹材前加水分解クラフトパルプの完全無塩素漂 白のためのモノ過硫酸処理に関する研究 学位授与大学 筑波大学 (University of Tsukuba) 学位授与年度 2015 報告番号 12102甲第7760号 URL http://hdl.handle.net/2241/00143908 Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps January 2016 Andri Taufick RIZALUDDIN Studies on Peroxymonosulfuric Acid Treatment for Totally Chlorine-free Bleaching of Hardwoods Prehydrolysis-kraft Pulps A Dissertation Submitted to The Graduate School of Life and Environmental Sciences, The University of Tsukuba In Partial Fulfillment on the Requirements For the Degree of Doctor of Philosophy in Bioresource Engineering (Doctoral Program in Appropriate Technology and Sciences for Sustainable Development) Andri Taufick RIZALUDDIN Table of Contents Chapter 1 Introduction ........................................................................................................... 1 1.1 Indonesian pulp and paper industry........................................................................... 1 1.2 Indonesia’s environmental policies regarding the pulp and paper industry .............. 2 1.3 Bleaching process .................................................................................................... 11 1.3.1 Oxygen bleaching .............................................................................................. 11 1.3.2 Chlorine dioxide bleaching -

Quality Performs
QUALITY PERFORMS. Monopersulfate Compound General Technical Attributes. QUALITY WORKS. MONOPERSULFATE COMPOUND PRODUCT INFORMATION What is OxoneTM? Applications* Oxone™ monopersulfate compound is a white, granular, n Swimming pool shock oxidizer free-flowing peroxygen that provides powerful non-chlorine n Printed wiring board microetchant oxidation for a wide variety of industrial and consumer uses. n Repulping aid for wet-strength resin destruction n Odor control agent in wastewater treatment The active ingredient of Oxone™ is potassium peroxymono- n Bleach component in denture cleanser and laundry sulfate, KHSO5, commonly known as potassium monoper- formulations sulfate, which is present as a component of a triple salt with n Disinfectant active ingredient n the formula 2KHSO5•KHSO4•K2SO4 (pentapotassium bis- Other uses, where its combination of powerful oxidation (peroxymonosulphate) bis(sulphate), [CAS 70693-62-8]). and relative safe handling properties are of value *As with any product, use of Oxone™ in a given application must be tested (including field testing, etc.) by the user in advance to determine suitability. The oxidizing power of Oxone™ is derived from its peracid chemistry; it is the first neutralization salt of peroxymonosulfuric acid H2SO5 (also known as Caro’s acid). Figure 2: Effect of pH on Oxone™ Solution Stability Standard Potential (3 wt% Solution at 32°C [90°F]) 110The standard electrode potential (E°) of KHSO5 is given by 2 the following half cell reaction: 100 20 21C (0F) – + – 0 90HSO5 + 2H + 2e- HSO4 + H2O E = 1.85V 1 0The thermodynamic potential is high enough for many 10 0room temperature oxidations, including: 5 3C (9F) 60 0 n Halide to active halogen pH 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 0n Oxidation of reduced sulfur and nitrogen compounds n Cyanide to cyanate 40 If a decomposition is associated with high temperature, n Epoxidation of olefins decomposition of the constituent salts of Oxone™ may 30n Baeyer-Villiger oxidation of ketones generate sulfuric acid, sulfur dioxide, or sulfur trioxide. -

Recent Developments on the Origin and Nature of Reductive Sulfurous Off-Odours in Wine
fermentation Review Recent Developments on the Origin and Nature of Reductive Sulfurous Off-Odours in Wine Nikolaus Müller 1,* and Doris Rauhut 2 1 Silvanerweg 9, 55595 Wallhausen, Germany 2 Department of Microbiology and Biochemistry, Hochschule Geisenheim University, Von-Lade-Straße 1, 65366 Geisenheim, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-6706-913103 Received: 9 July 2018; Accepted: 3 August 2018; Published: 8 August 2018 Abstract: Reductive sulfurous off-odors are still one of the main reasons for rejecting wines by consumers. In 2008 at the International Wine Challenge in London, approximately 6% of the more than 10,000 wines presented were described as faulty. Twenty-eight percent were described as faulty because they presented “reduced characters” similar to those presented by “cork taint” and in nearly the same portion. Reductive off-odors are caused by low volatile sulfurous compounds. Their origin may be traced back to the metabolism of the microorganisms (yeasts and lactic acid bacteria) involved in the fermentation steps during wine making, often followed by chemical conversions. The main source of volatile sulfur compounds (VSCs) are precursors from the sulfate assimilation pathway (SAP, sometimes named as the “sulfate reduction pathway” SRP), used by yeast to assimilate sulfur from the environment and incorporate it into the essential sulfur-containing amino acids methionine and cysteine. Reductive off-odors became of increasing interest within the last few years, and the method to remove them by treatment with copper (II) salts (sulfate or citrate) is more and more questioned: The effectiveness is doubted, and after prolonged bottle storage, they reappear quite often.