Suppression of Neuronal Cholesterol Biosynthesis Impairs Brain Functions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Century Journals Project (CJP)
China National Knowledge Infrastructure: Century Journals Project (CJP) Complete Title List - Series E (at July, 2008) Please note: Titles are sorted by Series, then Language (with English titles appearing first) , then by Pinyin title in English phonetic order. Earliest Control Journal Title Language Title in Pinyin Glossing of Title Former Title Freq. ISSN # CN # Publisher Issue in Status URL Notes Code Series CNKI E 南京医科大学学报 (英文版) English Nanjing Yike Daxue Xuebao (Yingwenban) Journal of Nanjing Medical University Journal of Nan bimonthly 1007-4376 32-1443/R 南京医科大学 1994/01 Active http://china.eastview.com/kns50/Navi/Bridge.aspx?L NJYY E 上海第二医科大学学报 (外文版) English Shanghai Dier Yike Daxue Xuebao Journal of Shanghai Second Medical University Journal of Sha semiannual 1001-6686 31-1589/R 上海第二医科大学 1987/01 Active http://china.eastview.com/kns50/Navi/Bridge.aspx?L SHEI (Yingwenban) (Foreign Language Edition) E 生物医学与环境科学 (英文版) English Shengwu Yi Xue Yu Huan Jing Ke Xue Biomedical and Environmental Sciences Biomedical an bimonthly 0895-3988 11-2816/Q 中国预防医学科学 1989/01 Active http://china.eastview.com/kns50/Navi/Bridge.aspx?L SWYX (Yingwenban) E 生殖与避孕 (英文版) English Shengzhi Yu Biyun (Yingwenban) Journal of Reproduction and Contraception Reproduction quarterly 1001-7844 31-1555/R 上海市计划生育科 1994/01 Active http://china.eastview.com/kns50/Navi/Bridge.aspx?L SZBW E 世界针灸杂志 (英文版) English Shijie Zhenjiu Zazhi (Yingwenban) World Journal of Acupuncture-Moxibustion World Journa quarterly 1003-5257 11-2892/R 世界针灸学会联合会 1992/01 Active http://china.eastview.com/kns50/Navi/Bridge.aspx?L -

A Complete Collection of Chinese Institutes and Universities For
Study in China——All China Universities All China Universities 2019.12 Please download WeChat app and follow our official account (scan QR code below or add WeChat ID: A15810086985), to start your application journey. Study in China——All China Universities Anhui 安徽 【www.studyinanhui.com】 1. Anhui University 安徽大学 http://ahu.admissions.cn 2. University of Science and Technology of China 中国科学技术大学 http://ustc.admissions.cn 3. Hefei University of Technology 合肥工业大学 http://hfut.admissions.cn 4. Anhui University of Technology 安徽工业大学 http://ahut.admissions.cn 5. Anhui University of Science and Technology 安徽理工大学 http://aust.admissions.cn 6. Anhui Engineering University 安徽工程大学 http://ahpu.admissions.cn 7. Anhui Agricultural University 安徽农业大学 http://ahau.admissions.cn 8. Anhui Medical University 安徽医科大学 http://ahmu.admissions.cn 9. Bengbu Medical College 蚌埠医学院 http://bbmc.admissions.cn 10. Wannan Medical College 皖南医学院 http://wnmc.admissions.cn 11. Anhui University of Chinese Medicine 安徽中医药大学 http://ahtcm.admissions.cn 12. Anhui Normal University 安徽师范大学 http://ahnu.admissions.cn 13. Fuyang Normal University 阜阳师范大学 http://fynu.admissions.cn 14. Anqing Teachers College 安庆师范大学 http://aqtc.admissions.cn 15. Huaibei Normal University 淮北师范大学 http://chnu.admissions.cn Please download WeChat app and follow our official account (scan QR code below or add WeChat ID: A15810086985), to start your application journey. Study in China——All China Universities 16. Huangshan University 黄山学院 http://hsu.admissions.cn 17. Western Anhui University 皖西学院 http://wxc.admissions.cn 18. Chuzhou University 滁州学院 http://chzu.admissions.cn 19. Anhui University of Finance & Economics 安徽财经大学 http://aufe.admissions.cn 20. Suzhou University 宿州学院 http://ahszu.admissions.cn 21. -

Mukden Medical College Was Well Regarded, and in Reports That ‘Its Teachers and Students Fled
J R Coll Physicians Edinb 2006; 36:179–184 PAPER © 2006 Royal College of Physicians of Edinburgh Mukden Medical College (1911–1949): an outpost of Edinburgh medicine in northeast China. Part 2: 1918–1949: expansion, occupation, liberation and merger DS Crawford Emeritus Librarian, McGill University, Montréal, Québec, Canada; Honorary Research Librarian, China Medical University, Shenyang, China ABSTRACT Scottish physician Dugald Christie commenced practice in the city Correspondence to DS Crawford, of Mukden (Shenyang) in Manchuria in 1883. In 1911, he founded the Mukden 135 George Street South, Suite 304 Medical College, the first Western medical school in Manchuria. Edinburgh- Toronto, Ontario trained physicians and surgeons largely staffed the college and in 1934 it Canada M5A 4E8 became the first foreign university to have its medical degree recognised by the University of Edinburgh. It was merged into the China Medical University tel. +1 (416) 504 7636 (Zhongguo yi ke da xue) in 1949. During its separate existence the Mukden e-mail [email protected] Medical College brought modern medicine and medical education to northeastern China, and its legacy continues to influence both medical practice and medical education in China. KEYWORDS China Medical University, Chinese history, Dugald Christie, medical education, missions and missionaries, Mukden Medical College, University of Edinburgh LIST OF ABBREVIATIONS China Medical Board (CMB), China Medical Missionary Association (CMMA), Mukden Medical College (MMC), Peking Union Medical College (PUMC), Royal College of Physicians of Edinburgh (RCPE), Royal College of Surgeons of Edinburgh (RCSEd), South Manchuria Medical College (SMMC), United Nations Relief and Rehabilitation Administration (UNRRA) DECLARATION OF INTERESTS No conflict of interests declared. -

University of Leeds Chinese Accepted Institution List 2021
University of Leeds Chinese accepted Institution List 2021 This list applies to courses in: All Engineering and Computing courses School of Mathematics School of Education School of Politics and International Studies School of Sociology and Social Policy GPA Requirements 2:1 = 75-85% 2:2 = 70-80% Please visit https://courses.leeds.ac.uk to find out which courses require a 2:1 and a 2:2. Please note: This document is to be used as a guide only. Final decisions will be made by the University of Leeds admissions teams. -

Confidential: for Review Only
BMJ Confidential: For Review Only Incidence of type 1 diabetes mellitus in China: population based study, 2010 – 2013 Journal: BMJ Manuscript ID BMJ.2017.040488 Article Type: Research BMJ Journal: BMJ Date Submitted by the Author: 21-Jul-2017 Complete List of Authors: Weng, Jianping; Sun Yat-sen University Third Hospital; Guangdong Provincial Key Laboratory of Diabetology Zhou, Zhiguang; the Second Xiangya Hospital, Central South University, Institue of Endocrinology and Metabolism Guo, Lixin; Beijing Hospital Zhu, Dalong; Nanjing University Medical School Affiliated Nanjing Drum Tower Hospital Ji, Linong; Peking University People’s Hospital, Department of Endocrinology and Metabolism Luo, Xiaoping; Tongji Hospital of Tongji Medical College, Huazhong University of Science & Technology Mu, Yiming; People’s Liberation Army General Hospital, China, Jia, Wei-Ping; The Sixth Affiliated People’s Hospital, Shanghai Jiao Tong University, YANG, WEN YING; CHINA JAPAN FRIENDSHIP HOSPITAL, ENDOCRINOLOGY Kuang, Hongyu; The First Affiliated Hospital of Harbin Medical University Li, Qiang; The Second Affiliated Hospital of Harbin Medical University, Li, Yanbing; The First Affiliated Hospital of Sun Yat-sen University Yuan, Li; Union Hospital, Tongji Medical College, Huazhong University of Science & Technology Yu, Xuefeng; Wuhan Tongji Hospital, China, Shan, Zhongyan; First Affiliated Hospital, Chinese Medical University, Ji, Qiuhe; Xijing Hospital, Fourth Military Medical University, Ran, Xing-wu; West China Hospital of Sichuan University, Diabetic -
1 Please Read These Instructions Carefully
PLEASE READ THESE INSTRUCTIONS CAREFULLY. MISTAKES IN YOUR CSC APPLICATION COULD LEAD TO YOUR APPLICATION BEING REJECTED. Visit http://studyinchina.csc.edu.cn/#/login to CREATE AN ACCOUNT. • The online application works best with Firefox or Internet Explorer (11.0). Menu selection functions may not work with other browsers. • The online application is only available in Chinese and English. 1 • Please read this page carefully before clicking on the “Application online” tab to start your application. 2 • The Program Category is Type B. • The Agency No. matches the university you will be attending. See Appendix A for a list of the Chinese university agency numbers. • Use the + by each section to expand on that section of the form. 3 • Fill out your personal information accurately. o Make sure to have a valid passport at the time of your application. o Use the name and date of birth that are on your passport. Use the name on your passport for all correspondences with the CLIC office or Chinese institutions. o List Canadian as your Nationality, even if you have dual citizenship. Only Canadian citizens are eligible for CLIC support. o Enter the mailing address for where you want your admission documents to be sent under Permanent Address. Leave Current Address blank. Contact your home or host university coordinator to find out when you will receive your admission documents. Contact information for you home university CLIC liaison can be found here: http://clicstudyinchina.com/contact-us/ 4 • Fill out your Education and Employment History accurately. o For Highest Education enter your current degree studies. -

The Ethics Committees of All Participating Centers
Supplementary material BMJ Open The ethics committees of all participating centers The ethics committees of the First Affiliated Hospital of Dalian Medical University (Registration No: YJ-KY-2017-119) . The ethics committees of the Chinese PLA General Hospital (Registration No: S2018- 021-01). The ethics committees of the first Affiliated Hospital of Xi’an Jiaotong University (Registration No: XJTU1AF2018LSK-05). Research Ethics Committee of Guangdong General Hospital, Guangdong Academy of Medical Sciences (Registration No: GDREC2018001H [R1] ). The ethics committees of the Beijing Friendship Hospital, Capital Medical University (Registration No: 2018-P2-013-02). The ethics committees of the West China Hospital of Sichuan University (Registration No: 2018[39] ). The ethics committees of the Zhongshan Hospital, Fudan University (Registration No: B2018-109R). The ethics committees of the Dalian Municipal Central Hospital (Registration No: 2018-051-01). ICE for Clinical Research and Animal Trials of the First Affiliated Hospital, Sun Yat- sen University (Registration No:[2018]021). The ethics committees of the General Hospital of Ningxia Medical University (Registration No: 2018-137). Renji Hospital Ethics Committee of Shanghai Jiaotong University School of Medicine (Registration No: [2017]239). The ethics committees of the Huashan Hospital, Fudan University (Registration No:[2018]013). The ethics committees of the Xijing Hospital, The Fourth Military Medical University (Registration No: KY20182007-1). Chen J, et al. BMJ Open 2019; 9:e023162. doi: 10.1136/bmjopen-2018-023162 Supplementary material BMJ Open The ethics committees of the General Hospital of Benxi Iron and Steel Co., Ltd (Registration No: No applicable ). The ethics committees Daping Hospital Affiliated to Army Military Medical University (Registration No: [2018]20). -

World Bank Document
PHN Technical Notes GEN 15 Public Disclosure Authorized THE WORLD BANK MEDICAL EDUCATION IN CHINA Public Disclosure Authorized August 1983 Public Disclosure Authorized Population, iealth and Nutrition DIepartment This paper is one of a series issued by the Population, Health and Nutrition Department for the information and guidance of Bank staff working in these sectors. The views and opinions expressed in this paper do not necessarily reflect those of the Bank. Public Disclosure Authorized GEN 15 A B S T R A C T 1. Physicians play a critical role in shaping health policy and in health expenditure decisions. These decisions, while influenced.by the institutional environment of the physician, are also influenced by the physician' s previous education and by the 6pportunity to continue such education. Hence the quality of training provided by medical colleges greatly influences the effectiveness of re-irce allocation throughout the health care system. Moreover, because medical colleges constitute the peak of the medical hierarchy, the standards of professional ethics, skills and medical research that they uphold exert a profound influence on the entire medical community. In light of this effect, this paper examines the back- ground anid current status of medical education in China,.including plans for -medical manpower development and subject area focus.- It also discusses both subject and systems problems and issues in medical education, including gaps in the knowledge base, didactic. teaching and insufficient attention to health impact.of medical education. This paper is Supplementary Paper Number 6 to World Bank Report No. 4664-CHA, "The Health Sector in China." Prepared by John R. -
CONICYT Ranking Por Disciplina > Sub-Área OECD (Académicas) Comisión Nacional De Investigación 1
CONICYT Ranking por Disciplina > Sub-área OECD (Académicas) Comisión Nacional de Investigación 1. Ciencias Naturales > 1.3 Ciencias Físicas y Astronomía Científica y Tecnológica PAÍS INSTITUCIÓN RANKING PUNTAJE FRANCE Universite Paris Saclay (ComUE) 1 5,000 USA University of California Berkeley 2 5,000 USA California Institute of Technology 3 5,000 USA Massachusetts Institute of Technology (MIT) 4 5,000 USA Harvard University 5 5,000 USA Stanford University 6 5,000 UNITED KINGDOM University of Cambridge 7 5,000 FRANCE Sorbonne Universite 8 5,000 USA University of Chicago 9 5,000 JAPAN University of Tokyo 10 5,000 UNITED KINGDOM University of Oxford 11 5,000 FRANCE Universite Sorbonne Paris Cite-USPC (ComUE) 12 5,000 FRANCE University of Paris Diderot 13 5,000 FRANCE PSL Research University Paris (ComUE) 14 5,000 USA Princeton University 15 5,000 FRANCE Universite Paris Sud - Paris XI 16 5,000 CHINA Tsinghua University 17 5,000 USA University of Maryland College Park 18 5,000 UNITED KINGDOM University College London 19 5,000 UNITED KINGDOM Imperial College London 20 5,000 FRANCE Communaute Universite Grenoble Alpes 21 5,000 USA University of Michigan 22 5,000 CANADA University of Toronto 23 5,000 FRANCE Universite Grenoble Alpes (UGA) 24 5,000 ITALY Sapienza University Rome 25 5,000 ITALY University of Padua 26 5,000 CHINA Peking University 27 5,000 UNITED KINGDOM University of Edinburgh 28 5,000 USA University of Illinois Urbana-Champaign 29 5,000 USA Columbia University 30 5,000 INDIA Indian Institute of Technology System (IIT System) -

Shengjing Hospital of China Medical University's Success Story
SHENGJING HOSPITAL OF CHINA MEDICAL UNIVERSITY EQUIPS ITS CAMPUS WITH THE BIGGEST EXTERNAL WIRELESS NETWORK IN CHINESE HEALTHCARE SYSTEM THANKS TO ALCATEL-LUCENT SHENGJING HOSPITAL OF CHINA MEDICAL UNIVERSITY CUSTOMER INFORMATION • Shengjing hospital of China Medical University is the result of the merger of the former national Shenyang Medical College founded in 1911 and the former private Liaoning Medical College founded in 1892. • CMU includes 18 colleges, faculties and sections, 130 departments: all with numerous disciplines (specialties). They are authorized to grant doctor and master degrees; 11 specialties for undergraduates and 10 specialties for higher professional techniques. CHALLENGES • Needed to have a network and communication system (voice and contact center) suitable for future expansion since the customer wanted to avoid a short term investment • As this was the biggest wireless network in the Chinese healthcare system, it was important to make sure all areas had a functional wireless network, even if one was inside or outside the campus building, or inside a lift. The Wi-Fi is a basic communication requirement and is widely utilized within the campus. “The integrated network setup brings revolutionary change to the hospital. This includes a good information system and ability to maintain PRODUCTS, SOLUTIONS AND SERVICES a quality network that would otherwise not be fully utilized. If we didn’t • Alcatel-Lucent OmniSwitch™ 9700 Chassis LAN Switch set up Alcatel-Lucent’s integrated network, it would be very difficult to -
Table S1. Self-Rated Questionnaire Assessing the Eligibility of Ascam Hospitals
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Stroke Vasc Neurol Table S1. Self-rated questionnaire assessing the eligibility of ASCaM hospitals. Evaluation Serial Evaluation Evaluation factors Score Scoring standards items number method The hospital selected for the thrombolysis map should have a person responsible for View the thrombolysis project for acute ischemic personnel Deduct 3 points if no relevant 1 3.0 stroke who is also responsible for registration personnel are identified. coordinating communication between the status ) project and the emergency center. 20 Set up a special reception telephone and set it to record in the prehospital emergency dispatching command system to ensure smooth 24-hour calls, and designated staff Field members are responsible for answering and inspection of Deduct 1 point for each missing 2 recording calls. If a related WeChat public 3.0 equipment and item until the score is zero. platform, mobile APP or intelligent digital staffing monitoring transmission system is available, the hospital must guarantee that anyone can reply immediately at any time within 24 hours. In case of inadequate or loss of medical capacity due to special circumstances (e.g., CT failure), the hospital's general-on-duty Field or project leader must inform the EMS inspection and Deduct 3 points for no relevant 3 3.0 command center by fax or dedicated regular spot capacity. landline in advance. The hospital must checks Connection between prehospital between and Connection in-hospital( accept (suspected) cerebrovascular disease patients who are transferred by EMS 24/7. -
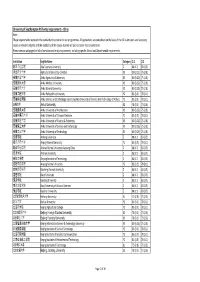
University of Southampton PGT Entry Requirements ‐ China Note: These Requirements Represent the Standard Entry Criteria for Our Programmes
University of Southampton PGT entry requirements ‐ China Note: These requirements represent the standard entry criteria for our programmes. All applications are considered on the basis of the full submission, and so strong scores in relevant subjects and the suitability of the course studied will also be taken into consideration. Please see our webpages for full information and entry requirements, including specific School and Departmental requirements. Institution English Name Category 2:1 2:2 阿坝师范学院 Aba Teachers University Z 88 (3.7) 83 (3.5) 河北农业大学 Agricultural University of Hebei X3 80 (3.25) 75 (2.8) 安徽农业大学 Anhui Agricultural University X3 80 (3.25) 75 (2.8) 安徽医科大学 Anhui Medical University X3 80 (3.25) 75 (2.8) 安徽师范大学 Anhui Normal University X3 80 (3.25) 75 (2.8) 安徽工程大学 Anhui Polytechnic University Y2 83 (3.5) 78 (3.1) 安徽科技学院 Anhui Science and Technology University (AKA University of Science And Technology of Anhui) Y2 83 (3.5) 78 (3.1) 安徽大学 Anhui University X2 78 (3.1) 73 (2.6) 安徽建筑大学 Anhui University of Architecture X3 80 (3.25) 75 (2.8) 安徽中医药大学 Anhui University of Chinese Medicine Y2 83 (3.5) 78 (3.1) 安徽财经大学 Anhui University of Finance & Economics X3 80 (3.25) 75 (2.8) 安徽理工大学 Anhui University of Science and Technology X3 80 (3.25) 75 (2.8) 安徽工业大学 Anhui University of Technology X3 80 (3.25) 75 (2.8) 安康学院 Ankang University Z 88 (3.7) 83 (3.5) 安庆师范大学 Anqing Normal University Y2 83 (3.5) 78 (3.1) 鞍山师范学院 Anshan Normal University Liaoning China Z 88 (3.7) 83 (3.5) 安顺学院 Anshun University Z 88 (3.7) 83 (3.5) 安阳工学院 Anyang Institute of Technology