Transcriptome-Wide Map of M6a Circrnas Identified in a Rat Model Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development
International Journal of Molecular Sciences Article Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development Nashwa El Hadidy 1 and Vladimir N. Uversky 1,2,* 1 Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC07, Tampa, FL 33612, USA; [email protected] 2 Laboratory of New Methods in Biology, Institute for Biological Instrumentation of the Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 Received: 20 September 2019; Accepted: 21 October 2019; Published: 23 October 2019 Abstract: The two-meter-long DNA is compressed into chromatin in the nucleus of every cell, which serves as a significant barrier to transcription. Therefore, for processes such as replication and transcription to occur, the highly compacted chromatin must be relaxed, and the processes required for chromatin reorganization for the aim of replication or transcription are controlled by ATP-dependent nucleosome remodelers. One of the most highly studied remodelers of this kind is the BRG1- or BRM-associated factor complex (BAF complex, also known as SWItch/sucrose non-fermentable (SWI/SNF) complex), which is crucial for the regulation of gene expression and differentiation in eukaryotes. Chromatin remodeling complex BAF is characterized by a highly polymorphic structure, containing from four to 17 subunits encoded by 29 genes. The aim of this paper is to provide an overview of the role of BAF complex in chromatin remodeling and also to use literature mining and a set of computational and bioinformatics tools to analyze structural properties, intrinsic disorder predisposition, and functionalities of its subunits, along with the description of the relations of different BAF complex subunits to the pathogenesis of various human diseases. -

A High Throughput, Functional Screen of Human Body Mass Index GWAS Loci Using Tissue-Specific Rnai Drosophila Melanogaster Crosses Thomas J
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. Baranski Washington University School of Medicine in St. Louis Aldi T. Kraja Washington University School of Medicine in St. Louis Jill L. Fink Washington University School of Medicine in St. Louis Mary Feitosa Washington University School of Medicine in St. Louis Petra A. Lenzini Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Baranski, Thomas J.; Kraja, Aldi T.; Fink, Jill L.; Feitosa, Mary; Lenzini, Petra A.; Borecki, Ingrid B.; Liu, Ching-Ti; Cupples, L. Adrienne; North, Kari E.; and Province, Michael A., ,"A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses." PLoS Genetics.14,4. e1007222. (2018). https://digitalcommons.wustl.edu/open_access_pubs/6820 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Thomas J. Baranski, Aldi T. Kraja, Jill L. Fink, Mary Feitosa, Petra A. Lenzini, Ingrid B. Borecki, Ching-Ti Liu, L. Adrienne Cupples, Kari E. North, and Michael A. Province This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6820 RESEARCH ARTICLE A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. -

The Loss of TET2 Promotes CD8+ T Cell Memory Differentiation Shannon A
The Loss of TET2 Promotes CD8+ T Cell Memory Differentiation Shannon A. Carty, Mercy Gohil, Lauren B. Banks, Renee M. Cotton, Matthew E. Johnson, Erietta Stelekati, Andrew D. This information is current as Wells, E. John Wherry, Gary A. Koretzky and Martha S. of September 29, 2021. Jordan J Immunol published online 17 November 2017 http://www.jimmunol.org/content/early/2017/11/17/jimmun ol.1700559 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2017/11/17/jimmunol.170055 Material 9.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 29, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published November 17, 2017, doi:10.4049/jimmunol.1700559 The Journal of Immunology The Loss of TET2 Promotes CD8+ T Cell Memory Differentiation Shannon A. Carty,*,1 Mercy Gohil,† Lauren B. Banks,† Renee M. Cotton,‡ Matthew E. Johnson,x Erietta Stelekati,{,‖ Andrew D. -

Mapping of M6a and Its Regulatory Targets in Prostate Cancer Reveals a METTL3-Low Induction of Therapy Resistance
Author Manuscript Published OnlineFirst on June 4, 2021; DOI: 10.1158/1541-7786.MCR-21-0014 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Mapping of m6A and Its Regulatory Targets in Prostate Cancer Reveals a METTL3-low Induction of Therapy Resistance Kellie A. Cotter1, John Gallon2, Nadine Uebersax1, Philip Rubin1, Kate D. Meyer3,4, Salvatore Piscuoglio2,5,6, Samie R. Jaffrey7*, Mark A. Rubin1,8,9,* 1 Department for BioMedical Research, University of Bern, Bern, Switzerland 2 Visceral surgery and Precision Medicine research laboratory, Department of Biomedicine, University of Basel, Basel, Switzerland 3 Department of Biochemisty, Duke University School of Medicine, Durham, NC, USA 4 Department of Neurobiology, Duke University School of Medicine, Durham, NC, USA 5 Institute of Medical Genetics and Pathology, University Hospital Basel, Basel, Switzerland 6 Clarunis, Department of Visceral Surgery, University Centre for Gastrointestinal and Liver Diseases, St. Clara Hospital and University Hospital Basel, Switzerland 7 Department of Pharmacology, Weill Cornell Medicine, New York, NY, USA 8 Inselspital, Bern, Switzerland 9 Bern Center for Precision Medicine, Bern, Switzerland * Co-senior and corresponding authors: [email protected] [email protected] RUNNING TITLE m6A targets in PCa: low METTL3 induces therapy resistance KEYWORDS m6A, epitranscriptomics, prostate cancer, METTL3, castration resistance FINANCIAL SUPPORT This project was supported by funding from the Prostate Cancer Foundation (18YOUN06, K.A.C.), the Weill Cornell Medicine SPORE in Prostate Cancer (Developmental Research Project, S.R.J. and M.A.R.), the Swiss Cancer League (KFS-4988-02-2020-R, S.P.), and the NIH (R01CA186702, S.R.J.). -

Proteomics of Nucleocytoplasmic Partitioning
Available online at www.sciencedirect.com ScienceDirect Proteomics of nucleocytoplasmic partitioning Thao Nguyen, Nishant Pappireddi and Martin Wu¨ hr The partitioning of the proteome between nucleus and activation. Kinase activity can also be regulated via sub- cytoplasm affects nearly every aspect of eukaryotic biology. cellular localization: cyclin B needs to relocalize into the Despite this central role, we still have a poor understanding of nucleus to induce NE breakdown, which is required for which proteins localize in the nucleus and how this varies in the transition from G2 to mitosis [3]. Considering the different cell types and conditions. Recent advances in importance of subcellular localization in encoding impor- quantitative proteomics and high-throughput imaging are tant cellular information, it is not surprising that mis- starting to close this knowledge gap. Studies on protein regulation of nuclear transport has been associated with interaction are beginning to reveal the spectrum of cargos of multiple diseases, including developmental defects and nuclear import and export receptors.We anticipate that it will cancer [4–6]. Targeting mis-regulation of NC partitioning soon be possible to predict each protein’s nucleocytoplasmic has emerged as a promising therapeutic approach, partic- localization based on its importin/exportin interactions and its ularly for cancer treatment [7–11]. estimated diffusion rate through the nuclear pore. This insight is likely to provide us with a fundamental understanding of Many previous studies have reported this subcellular how cells use nucleocytoplasmic partitioning to encode and localization of individual proteins. Recent technological relay information. advances in methods such as mass spectrometry (MS) now allow us to look at the entire proteome at once. -

Tepzz 8Z6z54a T
(19) TZZ ZZ_T (11) EP 2 806 054 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.11.2014 Bulletin 2014/48 C40B 40/06 (2006.01) C12Q 1/68 (2006.01) C40B 30/04 (2006.01) C07H 21/00 (2006.01) (21) Application number: 14175049.7 (22) Date of filing: 28.05.2009 (84) Designated Contracting States: (74) Representative: Irvine, Jonquil Claire AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HGF Limited HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 140 London Wall PT RO SE SI SK TR London EC2Y 5DN (GB) (30) Priority: 28.05.2008 US 56827 P Remarks: •Thecomplete document including Reference Tables (62) Document number(s) of the earlier application(s) in and the Sequence Listing can be downloaded from accordance with Art. 76 EPC: the EPO website 09753364.0 / 2 291 553 •This application was filed on 30-06-2014 as a divisional application to the application mentioned (71) Applicant: Genomedx Biosciences Inc. under INID code 62. Vancouver, British Columbia V6J 1J8 (CA) •Claims filed after the date of filing of the application/ after the date of receipt of the divisional application (72) Inventor: Davicioni, Elai R.68(4) EPC). Vancouver British Columbia V6J 1J8 (CA) (54) Systems and methods for expression- based discrimination of distinct clinical disease states in prostate cancer (57) A system for expression-based discrimination of distinct clinical disease states in prostate cancer is provided that is based on the identification of sets of gene transcripts, which are characterized in that changes in expression of each gene transcript within a set of gene transcripts can be correlated with recurrent or non- recur- rent prostate cancer. -
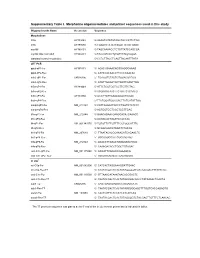
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Signatures of Adverse Pathological Features, Androgen Insensitivity and Metastatic Potential in Prostate Cancer
ANTICANCER RESEARCH 35: 5443-5452 (2015) Signatures of Adverse Pathological Features, Androgen Insensitivity and Metastatic Potential in Prostate Cancer YURI TOLKACH, AXEL MERSEBURGER, THOMAS HERRMANN, MARKUS KUCZYK, JÜRGEN SERTH and FLORIAN IMKAMP Clinic for Urology and Urologic Oncology, Hannover Medical School, Hannover, Germany Abstract. Background/Aim: The genetic characterization of The natural course of prostate cancer, its aggressiveness, prostate tumors is important for personalized therapy. The invasiveness and metastatic potential cannot be determined aim of the present study was to investigate the role of and predicted by clinical parameters alone (1, 2). Thus, genetic previously described prostate cancer-related genes in the characterization of prostate tumours is considered an essential genetic characterization of prostate tumors. Materials and step in the development of individualized therapy (3). All Methods: Forty-two genes were selected for expression tumors are separate entities, which, nevertheless, share some analysis (real time-quantitative polymerase chain reaction). common genetic changes (4). The identification of these One normal prostatic epithelial cell line and three would have a major clinical impact on everyday practice. standardized prostate cancer cell lines were used. Twenty- In recent years, multiple gene expression analysis (mRNA eight patients treated with radical prostatectomy were expression) of prostate cancer has been the focus of intensive included in the study. Results: The following genes appeared study. Previous attempts led to the identification of genes to be possibly related to the metastatic potential of the tumor: specific for high-grade tumours (5), aggressive prostate ELOVL fatty acid elongase 7 (ELOVL7), enhancer of zeste 2 cancer with a tendency towards recurrence (6-12), lethal polycomb repressive complex 2 subunit (EZH2), gastrulation tumours (10, 13) and hormone-refractory (14, 15) and brain homeobox 2 (GBX2), golgi membrane protein 1 metastatic prostate cancer (10, 16). -

A Deep Proteomics Perspective on CRM1-Mediated Nuclear Export and Nucleocytoplasmic Partitioning
A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning Koray Kırlı 1#, Samir Karaca1,2#, Heinz-Jürgen Dehne 1, Matthias Samwer 1,4, Kuan- Ting Pan 2, Christof Lenz 2,3, Henning Urlaub2,3* and Dirk Görlich1* 1 Department of Cellular Logistics and 2 Bioanalytical Mass Spectrometry Group, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany; 3 Bioanalytics, Institute for Clinical Chemistry, University Medical Center Göttingen, Göttingen, Germany. 4 Current address: Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA), Vienna, Austria. # Joint first authors; *Joint corresponding authors; E-mail: [email protected]; [email protected] Abstract CRM1 is a highly conserved, RanGTPase-driven exportin that carries proteins and RNPs from the nucleus to the cytoplasm. We now explored the cargo-spectrum of CRM1 in depth and identified surprisingly large numbers, namely >700 export substrates from the yeast S. cerevisiae, ≈ 1000 from Xenopus oocytes and >1050 from human cells. In addition, we quantified the partitioning of ≈5000 unique proteins between nucleus and cytoplasm of Xenopus oocytes. The data suggest new CRM1 functions in spatial control of vesicle coat-assembly, centrosomes, autophagy, peroxisome biogenesis, cytoskeleton, ribosome maturation, translation, mRNA degradation, and more generally in precluding a potentially detrimental action of cytoplasmic pathways within the nuclear interior. There are also numerous new instances where CRM1 appears to act in regulatory circuits. Altogether, our dataset allows unprecedented insights into the nucleocytoplasmic organisation of eukaryotic cells, into the contributions of an exceedingly promiscuous exportin and it provides a new basis for NES prediction. 2 Introduction The nuclear envelope (NE) separates the cell nucleus from the cytoplasm. -

Identification of Transcripts Overexpressed During Airway
ERJ Express. Published on March 5, 2008 as doi: 10.1183/09031936.00172107 IDENTIFICATION OF TRANSCRIPTS OVEREXPRESSED DURING AIRWAY EPITHELIUM DIFFERENTIATION Brigitte CHHIN (1, 2)*, Jacqueline PHAM (1, 2)*, Loubna EL ZEIN (1, 2), Karine KAISER (3), Olivier MERROT (4), Patrice BOUVAGNET (1, 2, 5) Laboratoire Cardiogénétique, EA 4171, Université Lyon 1, Lyon, France (1); Laboratoire Cardiogénétique, ERM 0107, INSERM, Lyon, France (2); Laboratoire de Parasitologie, Université Lyon 1, Lyon, France (3); Service ORL, Hôpital de la Croix-Rousse, Hospices Civils de Lyon, Lyon, France (4); Laboratoire Cardiogénétique, CBPE, Groupe Hospitalier Est, Hospices Civils de Lyon, Lyon, France (5). * These authors contributed equally to this study. Address for correspondence : Patrice BOUVAGNET Laboratoire Cardiogénétique, CBPE, Groupe Hospitalier Est 59, boulevard Pinel 69677 Bron, France Tel.: +33 472 12 96 76; Fax: +33 427 85 59 00 Mail: [email protected] Short title: AIRWAY EPITHELIUM SPECIFIC TRANSCRIPTS Copyright 2008 by the European Respiratory Society. ABSTRACT Human airway epithelium, the forefront defence protecting the respiratory tract, evacuates inhaled particles by a permanent beating of epithelial cells cilia. When deficient, this organelle causes Primary Ciliary Dyskinesia (PCD), and despite numerous studies, data regarding ciliated cells gene expression are still incomplete. To identify genes specifically expressed in human ciliated respiratory cells, we performed a transcriptional analysis. The transcriptome of de-differentiated epithelial cells was subtracted from fully re- differentiated cells using cDNA Representational Difference Analysis (RDA). To validate our results, gene overexpression in ciliated cells was confirmed by real-time PCR (RT-PCR), and by comparing our list of ciliated cell overexpressed genes to list obtained in previous studies.