Molecular, Evolutionary, and Structural Analysis of the Terminal Protein Domain of Hepatitis B Virus Polymerase, a Potential Drug Target
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Immunogenicity and Efficacy Testing in Chimpanzees of an Oral Hepatitis B Vaccine Based on Live Recombinant Adenovirus
Proc. Natl. Acad. Sci. USA Vol. 86, pp. 6763-6767, September 1989 Medical Sciences Immunogenicity and efficacy testing in chimpanzees of an oral hepatitis B vaccine based on live recombinant adenovirus (adenovirus animal model/adenovirus-vectored vacdnes) MICHAEL D. LUBECK*, ALAN R. DAVIS*, MURTY CHENGALVALA*, ROBERT J. NATUK*, JOHN E. MORIN*, KATHERINE MOLNAR-KIMBER*, BRUCE B. MASON*, BHEEM M. BHAT*, SATOSHI MIZUTANI*, PAUL P. HUNG*, AND ROBERT H. PURCELLO *Wyeth-Ayerst Research, Biotechnology and Microbiology Division, P.O. Pox 8299, Philadelphia, PA 19101; and tLaboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892 Contributed by Robert H. Purcell, May 30, 1989 ABSTRACT As a major cause of acute and chronic liver which, because oftheir wide prior usage, are good candidates disease as well as hepatocellular carcinoma, hepatitis B virus as vectors. Other less well-characterized small animal models (HBV) continues to pose significant health problems world- for human adenoviruses have been occasionally reported wide. Recombinant hepatitis B vaccines based on adenovirus (11-13) but are likewise nonpermissive for Ad4 and Ad7 vectors have been developed to address global needs for infections (unpublished data). An early study of animal effective control of hepatitits B infection. Although consider- species including nonhuman primates indicated that human able progress has been made in the construction ofrecombinant adenoviruses do not induce acute respiratory disease in adenoviruses that express large amounts of HBV gene prod- monkeys or chimpanzees following intranasal inoculations ucts, preclinical immunogenicity and efficacy testing of candi- (14). Serological data, however, indicated that such experi- date vaccines has remained difficult due to the lack ofa suitable mental infections occasionally induced anti-adenovirus anti- animal model. -

Hepatitis Virus in Long-Fingered Bats, Myanmar
DISPATCHES Myanmar; the counties are adjacent to Yunnan Province, Hepatitis Virus People’s Republic of China. The bats covered 6 species: Miniopterus fuliginosus (n = 640), Hipposideros armiger in Long-Fingered (n = 8), Rhinolophus ferrumequinum (n = 176), Myotis chi- nensis (n = 11), Megaderma lyra (n = 6), and Hipposideros Bats, Myanmar fulvus (n = 12). All bat tissue samples were subjected to vi- Biao He,1 Quanshui Fan,1 Fanli Yang, ral metagenomic analysis (unpublished data). The sampling Tingsong Hu, Wei Qiu, Ye Feng, Zuosheng Li, of bats for this study was approved by the Administrative Yingying Li, Fuqiang Zhang, Huancheng Guo, Committee on Animal Welfare of the Institute of Military Xiaohuan Zou, and Changchun Tu Veterinary, Academy of Military Medical Sciences, China. We used PCR to further study the prevalence of or- During an analysis of the virome of bats from Myanmar, thohepadnavirus in the 6 bat species; the condition of the a large number of reads were annotated to orthohepadnavi- samples made serologic assay and pathology impracticable. ruses. We present the full genome sequence and a morpho- Viral DNA was extracted from liver tissue of each of the logical analysis of an orthohepadnavirus circulating in bats. 853 bats by using the QIAamp DNA Mini Kit (QIAGEN, This virus is substantially different from currently known Hilden, Germany). To detect virus in the samples, we con- members of the genus Orthohepadnavirus and represents ducted PCR by using the TaKaRa PCR Kit (TaKaRa, Da- a new species. lian, China) with a pair of degenerate pan-orthohepadnavi- rus primers (sequences available upon request). The PCR he family Hepadnaviridae comprises 2 genera (Ortho- reaction was as follows: 45 cycles of denaturation at 94°C Thepadnavirus and Avihepadnavirus), and viruses clas- for 30 s, annealing at 54°C for 30 s, extension at 72°C for sified within these genera have a narrow host range. -

Hepatitis B Fast Facts Everything You Need to Know in 2 Minutes Or Less!
Hepatitis B Foundation Cause for a Cure www.hepb.org Hepatitis B Fast Facts Everything you need to know in 2 minutes or less! Hepatitis B is the most common serious liver infection in the world. It is caused by the hepatitis B virus (HBV) that attacks liver cells and can lead to liver failure, cirrhosis (scarring) or cancer of the liver. The virus is transmitted through contact with blood and bodily fluids that contain blood. Most people are able to fight off the hepatitis B infection and clear the virus from their blood. This may take up to six months. While the virus is present in their blood, infected people can pass the virus on to others. Approximately 5-10% of adults, 30-50% of children, and 90% of babies will not get rid of the virus and will develop chronic infection. Chronically infected people can pass the virus on to others and are at increased risk for liver problems later in life. The hepatitis B virus is 100 times more infectious than the AIDS virus. Yet, hepatitis B can be pre- vented with a safe and effective vaccine. For the 400 million people worldwide who are chronically infected with hepatitis B, the vaccine is of no use. However, there are promising new treatments for those who live with chronic hepatitis B. In the World: • This year alone, 10 to 30 million people will become infected with the hepatitis B virus (HBV). • The World Health Organization estimates that 400 million people worldwide are already chronically infected with hepatitis B. -

Prevention & Control of Viral Hepatitis Infection
Prevention & Control of Viral Hepatitis Infection: A Strategy for Global Action © World Health Organization 2011. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by WHO to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Table of contents Disease burden 02 What is viral hepatitis? 05 Prevention & control: a tailored approach 06 Global Achievements 08 Remaining challenges 10 World Health Assembly: a mandate for comprehensive prevention & control 13 WHO goals and strategy -

And Giant Guitarfish (Rhynchobatus Djiddensis)
VIRAL DISCOVERY IN BLUEGILL SUNFISH (LEPOMIS MACROCHIRUS) AND GIANT GUITARFISH (RHYNCHOBATUS DJIDDENSIS) BY HISTOPATHOLOGY EVALUATION, METAGENOMIC ANALYSIS AND NEXT GENERATION SEQUENCING by JENNIFER ANNE DILL (Under the Direction of Alvin Camus) ABSTRACT The rapid growth of aquaculture production and international trade in live fish has led to the emergence of many new diseases. The introduction of novel disease agents can result in significant economic losses, as well as threats to vulnerable wild fish populations. Losses are often exacerbated by a lack of agent identification, delay in the development of diagnostic tools and poor knowledge of host range and susceptibility. Examples in bluegill sunfish (Lepomis macrochirus) and the giant guitarfish (Rhynchobatus djiddensis) will be discussed here. Bluegill are popular freshwater game fish, native to eastern North America, living in shallow lakes, ponds, and slow moving waterways. Bluegill experiencing epizootics of proliferative lip and skin lesions, characterized by epidermal hyperplasia, papillomas, and rarely squamous cell carcinoma, were investigated in two isolated poopulations. Next generation genomic sequencing revealed partial DNA sequences of an endogenous retrovirus and the entire circular genome of a novel hepadnavirus. Giant Guitarfish, a rajiform elasmobranch listed as ‘vulnerable’ on the IUCN Red List, are found in the tropical Western Indian Ocean. Proliferative skin lesions were observed on the ventrum and caudal fin of a juvenile male quarantined at a public aquarium following international shipment. Histologically, lesions consisted of papillomatous epidermal hyperplasia with myriad large, amphophilic, intranuclear inclusions. Deep sequencing and metagenomic analysis produced the complete genomes of two novel DNA viruses, a typical polyomavirus and a second unclassified virus with a 20 kb genome tentatively named Colossomavirus. -

Hepatitis B? HEPATITIS B Hepatitis B Is a Contagious Liver Disease That Results from Infection with the Hepatitis B Virus
What is Hepatitis B? HEPATITIS B Hepatitis B is a contagious liver disease that results from infection with the Hepatitis B virus. When first infected, a person can develop Are you at risk? an “acute” infection, which can range in severity from a very mild illness with few or no symptoms to a serious condition requiring hospitalization. Acute Hepatitis B refers to the first 6 months after someone is exposed to the Hepatitis B virus. Some people are able to fight the infection and clear the virus. For others, the infection remains and leads to a “chronic,” or lifelong, illness. Chronic Hepatitis B refers to the illness that occurs when the Hepatitis B virus remains in a person’s body. Over time, the infection can cause serious health problems. How is Hepatitis B spread? Hepatitis B is usually spread when blood, semen, or other body fluids from a person infected with the Hepatitis B virus enter the body of someone who is not infected. This can happen through having sex with an infected partner; sharing needles, syringes, or other injection drug equipment; or from direct contact with the blood or open sores of an infected person. Hepatitis B can also be passed from an infected mother to her baby at birth. Who should be tested for Hepatitis B? Approximately 1.2 million people in the United States and 350 million people worldwide have Hepatitis B. Testing for Hepatitis B is recommended for certain groups of people, including: Most are unaware of their infection. ■ People born in Asia, Africa, and other regions with moderate or high rates Is Hepatitis B common? of Hepatitis B (see map) Yes. -
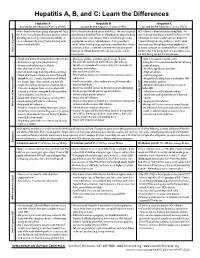
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Understanding Human Astrovirus from Pathogenesis to Treatment
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 6-2020 Understanding Human Astrovirus from Pathogenesis to Treatment Virginia Hargest University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Diseases Commons, Medical Sciences Commons, and the Viruses Commons Recommended Citation Hargest, Virginia (0000-0003-3883-1232), "Understanding Human Astrovirus from Pathogenesis to Treatment" (2020). Theses and Dissertations (ETD). Paper 523. http://dx.doi.org/10.21007/ etd.cghs.2020.0507. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Understanding Human Astrovirus from Pathogenesis to Treatment Abstract While human astroviruses (HAstV) were discovered nearly 45 years ago, these small positive-sense RNA viruses remain critically understudied. These studies provide fundamental new research on astrovirus pathogenesis and disruption of the gut epithelium by induction of epithelial-mesenchymal transition (EMT) following astrovirus infection. Here we characterize HAstV-induced EMT as an upregulation of SNAI1 and VIM with a down regulation of CDH1 and OCLN, loss of cell-cell junctions most notably at 18 hours post-infection (hpi), and loss of cellular polarity by 24 hpi. While active transforming growth factor- (TGF-) increases during HAstV infection, inhibition of TGF- signaling does not hinder EMT induction. However, HAstV-induced EMT does require active viral replication. -

In Vitro Selection and Characterization of Single Stranded DNA Aptamers Inhibiting the Hepatitis B Virus Capsid-Envelope Interaction
Aus dem Veterinärwissenschaftlichen Department der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München Arbeit angefertigt unter der Leitung von: Univ.-Prof. Dr. Gerd Sutter Angefertigt im Institut für Virologie des Helmholtz Zentrum München (apl.-Prof. Dr. Volker Bruss) In vitro Selection and Characterization of single stranded DNA Aptamers Inhibiting the Hepatitis B Virus Capsid-Envelope Interaction Inaugural-Dissertation zur Erlangung der tiermedizinischen Doktorwürde der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München von Ahmed El-Sayed Abd El-Halem Orabi aus Sharkia/Ägypten München 2013 Gedruckt mit der Genehmigung der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München Dekan: Univ.-Prof. Dr. Joachim Braun Berichterstatter: Univ.-Prof. Dr. Gerd Sutter Korreferent: Univ.-Prof. Dr. Bernd Kaspers Tag der Promotion: 20. Juli 2013 My Family Contents Contents 1 INTRODUCTION ................................................................................................... 1 2 REVIEW OF THE LITERATURE ....................................................................... 2 2.1 HEPATITIS B VIRUS (HBV) .............................................................................................. 2 2.1.1 HISTORY AND TAXONOMY .......................................................................................................... 2 2.1.2 EPIDEMIOLOGY AND PATHOGENESIS .......................................................................................... 3 2.1.3 VIRION STRUCTURE .................................................................................................................... -

Hepatitis B Factsheet: COVID-19 and Hep B
Hepatitis B factsheet: COVID-19 and hep B For more information about anything in this factsheet, phone the Hepatitis Infoline on 1800 803 990 or go to www.hep.org.au COVID-19 and hep B Frequently Asked Questions (FAQ) There’s so much information swirling around these days as we deal with the changing world around us due to COVID-19. There’s lots of incorrect information and it’s easy to be overwhelmed and confused. On this page, we’ll try to clear some things up. It is important that information is helpful, clear, and checked and confirmed by experts. Please share the following information – it comes from health and medical specialists. COVID-19 is an acronym that stands for COronaVIrus Disease 2019. You might hear it called coronavirus, corona, SARS-CoV-2, or even rona. We’ll call it COVID-19 or coronavirus in this FAQ but all these terms are essentially talking about the same thing and we’ll explain them at the bottom. You’ll also hear new or unfamiliar terms like “social distancing,” “physical distancing,” “self-isolation,” and “quarantine”. These terms and what they actually mean can become confusing so we’ve including a glossary at the bottom of this FAQ. This virus has only been known about since around December 2019 so it’s all very new, information changes quickly, and we’re all learning day-by-day and week-by-week. Compare COVID-19 to hep B which we’ve known about since the 1980s and hep B even earlier in the 1960s! We’ll regularly update this page as new information comes to light. -

Interaction Between Hepatitis B Virus and SARS-Cov-2 Infections
World Journal of W J G Gastroenterology Submit a Manuscript: https://www.f6publishing.com World J Gastroenterol 2021 March 7; 27(9): 782-793 DOI: 10.3748/wjg.v27.i9.782 ISSN 1007-9327 (print) ISSN 2219-2840 (online) MINIREVIEWS Interaction between hepatitis B virus and SARS-CoV-2 infections Tian-Dan Xiang, Xin Zheng ORCID number: Tian-Dan Xiang Tian-Dan Xiang, Xin Zheng, Department of Infectious Diseases, Union Hospital, Tongji Medical 0000-0003-2792-6631; Xin Zheng College, Huazhong University of Science and Technology, Wuhan 430022, Hubei Province, 0000-0001-6564-7807. China Author contributions: Zheng X Corresponding author: Xin Zheng, MD, PhD, Professor, Department of Infectious Diseases, contributed to the conception and Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. design of the work and revised the 1277 Jiefang Avenue, Wuhan 430022, Hubei Province, China. [email protected] manuscript; Xiang TD performed the literature review and drafted the manuscript. Abstract Conflict-of-interest statement: Coronavirus disease 2019 (COVID-19) has become a global pandemic and Authors declare no conflict of garnered international attention. The causative pathogen of COVID-19 is severe interest for this article. acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel, highly contagious coronavirus. Numerous studies have reported that liver injury is quite Open-Access: This article is an common in patients with COVID-19. Hepatitis B has a worldwide distribution as open-access article that was well as in China. At present, hepatitis B virus (HBV) remains a leading cause of selected by an in-house editor and cirrhosis, liver failure, and hepatocellular carcinoma. -

Hepatitis B Virus (HBV)
HEPATITIS B AND COLLEGE STUDENTS Hepatitis B is a liver disease that results from infection with the Hepatitis B virus (HBV). It can range in severity from a mild illness lasting a few weeks to a serious, lifelong illness. Hepatitis B is usually spread when blood, semen, or another body fluid from a person infected with the Hepatitis B virus enters the body of someone who is not infected. This can happen through sexual contact with an infected person or sharing needles, syringes, or other drug-injection equipment. Hepatitis B can also be passed from an infected mother to her baby at birth. Hepatitis B can be either acute or chronic. Acute HBV infection is a short-term illness that occurs within the first 6 months after someone is exposed to the Hepatitis B virus. Acute infection can — but does not always — lead to chronic infection. Chronic HBV infection is a long-term illness that occurs when the Hepatitis B virus remains in a person’s body. Chronic HBV is a serious disease that can result in long-term health problems, and even death. The best way to prevent HBV infection is by getting vaccinated. How common is hepatitis B in the United States? About 800,000 to 1.4 million persons in the United States have chronic HBV infection. Each year 38,000 more people, mostly young adults, get infected with HBV and almost 2,000 people die from chronic HBV. How is hepatitis B spread? Hepatitis B is spread when blood, semen, or other body fluid infected with the hepatitis B virus enters the body of a person who is not infected.