Hepatitis Virus in Long-Fingered Bats, Myanmar
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Virus Particle Structures
Virus Particle Structures Virus Particle Structures Palmenberg, A.C. and Sgro, J.-Y. COLOR PLATE LEGENDS These color plates depict the relative sizes and comparative virion structures of multiple types of viruses. The renderings are based on data from published atomic coordinates as determined by X-ray crystallography. The international online repository for 3D coordinates is the Protein Databank (www.rcsb.org/pdb/), maintained by the Research Collaboratory for Structural Bioinformatics (RCSB). The VIPER web site (mmtsb.scripps.edu/viper), maintains a parallel collection of PDB coordinates for icosahedral viruses and additionally offers a version of each data file permuted into the same relative 3D orientation (Reddy, V., Natarajan, P., Okerberg, B., Li, K., Damodaran, K., Morton, R., Brooks, C. and Johnson, J. (2001). J. Virol., 75, 11943-11947). VIPER also contains an excellent repository of instructional materials pertaining to icosahedral symmetry and viral structures. All images presented here, except for the filamentous viruses, used the standard VIPER orientation along the icosahedral 2-fold axis. With the exception of Plate 3 as described below, these images were generated from their atomic coordinates using a novel radial depth-cue colorization technique and the program Rasmol (Sayle, R.A., Milner-White, E.J. (1995). RASMOL: biomolecular graphics for all. Trends Biochem Sci., 20, 374-376). First, the Temperature Factor column for every atom in a PDB coordinate file was edited to record a measure of the radial distance from the virion center. The files were rendered using the Rasmol spacefill menu, with specular and shadow options according to the Van de Waals radius of each atom. -

Discovery of a Highly Divergent Hepadnavirus in Shrews from China
Virology 531 (2019) 162–170 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Discovery of a highly divergent hepadnavirus in shrews from China T Fang-Yuan Niea,b,1, Jun-Hua Tianc,1, Xian-Dan Lind,1, Bin Yuc, Jian-Guang Xinge, Jian-Hai Caof, ⁎ ⁎⁎ Edward C. Holmesg,h, Runlin Z. Maa, , Yong-Zhen Zhangb,h, a State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China b State Key Laboratory for Infectious Disease Prevention and Control; Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases; Department of Zoonoses, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Changping, Beijing, China c Wuhan Center for Disease Control and Prevention, Wuhan, China d Wenzhou Center for Disease Control and Prevention, Wenzhou, Zhejiang Province, China e Wencheng Center for Disease Control and Prevention, Wencheng, Zhejiang Province, China f Longwan Center for Disease Control and Prevention, Longwan District, Wenzhou, Zhejiang Province, China g Marie Bashir Institute for Infectious Diseases and Biosecurity, Charles Perkins Centre, School of Life and Environmental Sciences and Sydney Medical School, The University of Sydney, Sydney, NSW 2006, Australia h Shanghai Public Health Clinical Center & Institute of Biomedical Sciences, Fudan University, Shanghai, China ARTICLE INFO ABSTRACT Keywords: Limited sampling means that relatively little is known about the diversity and evolutionary history of mam- Hepadnaviruses malian members of the Hepadnaviridae (genus Orthohepadnavirus). An important case in point are shrews, the Shrews fourth largest group of mammals, but for which there is limited knowledge on the role they play in viral evo- Phylogeny lution and emergence. -

And Giant Guitarfish (Rhynchobatus Djiddensis)
VIRAL DISCOVERY IN BLUEGILL SUNFISH (LEPOMIS MACROCHIRUS) AND GIANT GUITARFISH (RHYNCHOBATUS DJIDDENSIS) BY HISTOPATHOLOGY EVALUATION, METAGENOMIC ANALYSIS AND NEXT GENERATION SEQUENCING by JENNIFER ANNE DILL (Under the Direction of Alvin Camus) ABSTRACT The rapid growth of aquaculture production and international trade in live fish has led to the emergence of many new diseases. The introduction of novel disease agents can result in significant economic losses, as well as threats to vulnerable wild fish populations. Losses are often exacerbated by a lack of agent identification, delay in the development of diagnostic tools and poor knowledge of host range and susceptibility. Examples in bluegill sunfish (Lepomis macrochirus) and the giant guitarfish (Rhynchobatus djiddensis) will be discussed here. Bluegill are popular freshwater game fish, native to eastern North America, living in shallow lakes, ponds, and slow moving waterways. Bluegill experiencing epizootics of proliferative lip and skin lesions, characterized by epidermal hyperplasia, papillomas, and rarely squamous cell carcinoma, were investigated in two isolated poopulations. Next generation genomic sequencing revealed partial DNA sequences of an endogenous retrovirus and the entire circular genome of a novel hepadnavirus. Giant Guitarfish, a rajiform elasmobranch listed as ‘vulnerable’ on the IUCN Red List, are found in the tropical Western Indian Ocean. Proliferative skin lesions were observed on the ventrum and caudal fin of a juvenile male quarantined at a public aquarium following international shipment. Histologically, lesions consisted of papillomatous epidermal hyperplasia with myriad large, amphophilic, intranuclear inclusions. Deep sequencing and metagenomic analysis produced the complete genomes of two novel DNA viruses, a typical polyomavirus and a second unclassified virus with a 20 kb genome tentatively named Colossomavirus. -
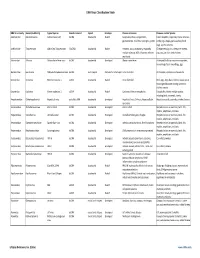
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

In Vitro Selection and Characterization of Single Stranded DNA Aptamers Inhibiting the Hepatitis B Virus Capsid-Envelope Interaction
Aus dem Veterinärwissenschaftlichen Department der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München Arbeit angefertigt unter der Leitung von: Univ.-Prof. Dr. Gerd Sutter Angefertigt im Institut für Virologie des Helmholtz Zentrum München (apl.-Prof. Dr. Volker Bruss) In vitro Selection and Characterization of single stranded DNA Aptamers Inhibiting the Hepatitis B Virus Capsid-Envelope Interaction Inaugural-Dissertation zur Erlangung der tiermedizinischen Doktorwürde der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München von Ahmed El-Sayed Abd El-Halem Orabi aus Sharkia/Ägypten München 2013 Gedruckt mit der Genehmigung der Tierärztlichen Fakultät der Ludwig-Maximilians-Universität München Dekan: Univ.-Prof. Dr. Joachim Braun Berichterstatter: Univ.-Prof. Dr. Gerd Sutter Korreferent: Univ.-Prof. Dr. Bernd Kaspers Tag der Promotion: 20. Juli 2013 My Family Contents Contents 1 INTRODUCTION ................................................................................................... 1 2 REVIEW OF THE LITERATURE ....................................................................... 2 2.1 HEPATITIS B VIRUS (HBV) .............................................................................................. 2 2.1.1 HISTORY AND TAXONOMY .......................................................................................................... 2 2.1.2 EPIDEMIOLOGY AND PATHOGENESIS .......................................................................................... 3 2.1.3 VIRION STRUCTURE .................................................................................................................... -

The Approved List of Biological Agents Advisory Committee on Dangerous Pathogens Health and Safety Executive
The Approved List of biological agents Advisory Committee on Dangerous Pathogens Health and Safety Executive © Crown copyright 2021 First published 2000 Second edition 2004 Third edition 2013 Fourth edition 2021 You may reuse this information (excluding logos) free of charge in any format or medium, under the terms of the Open Government Licence. To view the licence visit www.nationalarchives.gov.uk/doc/ open-government-licence/, write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email [email protected]. Some images and illustrations may not be owned by the Crown so cannot be reproduced without permission of the copyright owner. Enquiries should be sent to [email protected]. The Control of Substances Hazardous to Health Regulations 2002 refer to an ‘approved classification of a biological agent’, which means the classification of that agent approved by the Health and Safety Executive (HSE). This list is approved by HSE for that purpose. This edition of the Approved List has effect from 12 July 2021. On that date the previous edition of the list approved by the Health and Safety Executive on the 1 July 2013 will cease to have effect. This list will be reviewed periodically, the next review is due in February 2022. The Advisory Committee on Dangerous Pathogens (ACDP) prepares the Approved List included in this publication. ACDP advises HSE, and Ministers for the Department of Health and Social Care and the Department for the Environment, Food & Rural Affairs and their counterparts under devolution in Scotland, Wales & Northern Ireland, as required, on all aspects of hazards and risks to workers and others from exposure to pathogens. -

Orthohepadnavirus Avihepadnavirus
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the separate document “Help with completing a taxonomic proposal” Please try to keep related proposals within a single document; you can copy the modules to create more than one genus within a new family, for example. MODULE 1: TITLE, AUTHORS, etc (to be completed by ICTV Code assigned: 2016.019aD officers) Short title: One new species in the genus Orthohepadnavirus (e.g. 6 new species in the genus Zetavirus) Modules attached 2 3 4 5 (modules 1 and 10 are required) 6 7 8 9 10 Author(s): Heléne Norder and Lars Magnius Corresponding author with e-mail address: [email protected] List the ICTV study group(s) that have seen this proposal: A list of study groups and contacts is provided at http://www.ictvonline.org/subcommittees.asp . If Hepadnaviridae & Hepatitis delta virus Study in doubt, contact the appropriate subcommittee chair (fungal, invertebrate, plant, prokaryote or Group vertebrate viruses) ICTV Study Group comments (if any) and response of the proposer: Date first submitted to ICTV: July 12, 2016 Date of this revision (if different to above): August 2, 2016 ICTV-EC comments and response of the proposer: Decision: Response: Page 1 of 5 MODULE 2: NEW SPECIES creating and naming one or more new species. If more than one, they should be a group of related species belonging to the same genus. All new species must be placed in a higher taxon. -

New Avian Hepadnavirus in Palaeognathous Bird, Germany
RESEARCH LETTERS New Avian Hepadnavirus includes emus (Dromaius novaehollandiae) and ostrich- es (Struthio spp.). in Palaeognathous Bird, In 2015, a deceased adult elegant-crested tinamou Germany kept at Wuppertal Zoo (Wuppertal, Germany) underwent necropsy at the University of Veterinary Medicine Han- 1 1 nover, Foundation (Hannover, Germany). Initial histologic Wendy K. Jo, Vanessa M. Pfankuche, examination revealed moderate, necrotizing hepatitis and Henning Petersen, Samuel Frei, Maya Kummrow, inclusion body–like structures within the hepatocytes. To Stephan Lorenzen, Martin Ludlow, Julia Metzger, identify a putative causative agent, we isolated nucleic ac- Wolfgang Baumgärtner, Albert Osterhaus, ids from the liver and prepared them for sequencing on an Erhard van der Vries Illumina MiSeq system (Illumina, San Diego, CA, USA) Author affiliations: University of Veterinary Medicine Hannover, (online Technical Appendix, https://wwwnc.cdc.gov/EID/ Foundation, Hannover, Germany (W.K. Jo, V.M. Pfankuche, article/23/12/16-1634-Techapp1.pdf). We compared ob- H. Petersen, M. Ludlow, J. Metzger, W. Baumgärtner, tained reads with sequences in GenBank using an in-house A. Osterhaus, E. van der Vries); Center for Systems metagenomics pipeline. Approximately 78% of the reads Neuroscience, Hannover (W.K. Jo, V.M. Pfankuche, aligned to existing avihepadnavirus sequences. A full ge- W. Baumgärtner, A. Osterhaus); Wuppertal Zoo, Wuppertal, nome (3,024 bp) of the putative elegant-crested tinamou Germany (S. Frei, M. Kummrow); Bernhard Nocht Institute for HBV (ETHBV) was subsequently constructed by de novo Tropical Medicine, Hamburg (S. Lorenzen); Artemis One Health, assembly mapping >2 million reads (88.6%) to the virus Utrecht, the Netherlands (A. Osterhaus) genome (GenBank accession no. -

Packaging of Genomic RNA in Positive-Sense Single-Stranded RNA Viruses: a Complex Story
viruses Review Packaging of Genomic RNA in Positive-Sense Single-Stranded RNA Viruses: A Complex Story Mauricio Comas-Garcia 1,2 1 Research Center for Health Sciences and Biomedicine (CICSaB), Universidad Autónoma de San Luis Potosí (UASLP), Av. Sierra Leona 550 Lomas 2da Seccion, 72810 San Luis Potosi, Mexico; [email protected] 2 Department of Sciences, Universidad Autónoma de San Luis Potosí (UASLP), Av. Chapultepec 1570, Privadas del Pedregal, 78295 San Luis Potosi, Mexico Received: 14 February 2019; Accepted: 8 March 2019; Published: 13 March 2019 Abstract: The packaging of genomic RNA in positive-sense single-stranded RNA viruses is a key part of the viral infectious cycle, yet this step is not fully understood. Unlike double-stranded DNA and RNA viruses, this process is coupled with nucleocapsid assembly. The specificity of RNA packaging depends on multiple factors: (i) one or more packaging signals, (ii) RNA replication, (iii) translation, (iv) viral factories, and (v) the physical properties of the RNA. The relative contribution of each of these factors to packaging specificity is different for every virus. In vitro and in vivo data show that there are different packaging mechanisms that control selective packaging of the genomic RNA during nucleocapsid assembly. The goals of this article are to explain some of the key experiments that support the contribution of these factors to packaging selectivity and to draw a general scenario that could help us move towards a better understanding of this step of the viral infectious cycle. Keywords: (+)ssRNA viruses; RNA packaging; virion assembly; packaging signals; RNA replication 1. Introduction Nucleocapsid assembly and the RNA replication of positive-sense single-stranded RNA [(+)ssRNA] viruses occur in the cytoplasm. -

Complete Genome Sequence of a Divergent Strain of Tibetan Frog Hepatitis B Virus Associated to Concave-Eared Torrent Frog
bioRxiv preprint doi: https://doi.org/10.1101/506550; this version posted December 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Title: Complete genome sequence of a divergent strain of Tibetan frog hepatitis B virus 2 associated to concave-eared torrent frog Odorrana tormota 3 4 Humberto J. Debat 1*, Terry Fei Fan Ng2 5 6 1Instituto de Patología Vegetal, Centro de Investigaciones Agropecuarias, Instituto Nacional de 7 Tecnología Agropecuaria (IPAVE-CIAP-INTA), X5020ICA, Córdoba, Argentina. 8 2College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA 30602. 9 10 Running title: Tibetan frog hepatitis B virus in Odorrana tormota 11 12 *Address correspondence to: 13 Humberto J. Debat [email protected] 14 15 ORCID ID: 16 HJD 0000-0003-3056-3739, TFFN 0000-0002-4815-8697 17 18 Keywords: dsDNA virus, Hepadnavirus, amphibian virology, frog virus, Odorrana tormota, virus 19 discovery. 20 21 Abstract 22 The family Hepadnaviridae is characterized by partially dsDNA circular viruses of approximately 3.2 kb, 23 which are reverse transcribed from RNA intermediates. Hepadnaviruses (HBVs) have a broad host range 24 which includes humans (Hepatitis B virus), other mammals (genus Orthohepadnavirus), and birds 25 (Avihepadnavirus). HBVs host specificity has been expanded by reports of new viruses infecting fish, 26 amphibians, and reptiles. The tibetan frog hepatitis B virus (TFHBV) was recently discovered in 27 Nanorana parkeri (Family Dicroglossidae) from Tibet. -

BMBL) Quickly Became the Cornerstone of Biosafety Practice and Policy in the United States Upon First Publication in 1984
Biosafety in Microbiological and Biomedical Laboratories 5th Edition U.S. Department of Health and Human Services Public Health Service Centers for Disease Control and Prevention National Institutes of Health HHS Publication No. (CDC) 21-1112 Revised December 2009 Foreword Biosafety in Microbiological and Biomedical Laboratories (BMBL) quickly became the cornerstone of biosafety practice and policy in the United States upon first publication in 1984. Historically, the information in this publication has been advisory is nature even though legislation and regulation, in some circumstances, have overtaken it and made compliance with the guidance provided mandatory. We wish to emphasize that the 5th edition of the BMBL remains an advisory document recommending best practices for the safe conduct of work in biomedical and clinical laboratories from a biosafety perspective, and is not intended as a regulatory document though we recognize that it will be used that way by some. This edition of the BMBL includes additional sections, expanded sections on the principles and practices of biosafety and risk assessment; and revised agent summary statements and appendices. We worked to harmonize the recommendations included in this edition with guidance issued and regulations promulgated by other federal agencies. Wherever possible, we clarified both the language and intent of the information provided. The events of September 11, 2001, and the anthrax attacks in October of that year re-shaped and changed, forever, the way we manage and conduct work -

A Novel Orthohepadnavirus Identified in a Dead Maxwell's Duiker
viruses Communication A Novel Orthohepadnavirus Identified in a Dead Maxwell’s Duiker (Philantomba maxwellii) in Taï National Park, Côte d’Ivoire Jan F. Gogarten 1,2,3,4 , Markus Ulrich 1, Nishit Bhuva 3, Joel Garcia 3, Komal Jain 3, Bohyun Lee 3, Therese Löhrich 1,5, Alexandra Oleynik 3, Emmanuel Couacy-Hymann 6, Terence Fuh Neba 5, Nischay Mishra 3, Thomas Briese 3,*,†,Sébastien Calvignac-Spencer 1,2,*,†, W. Ian Lipkin 3,*,† and Fabian H. Leendertz 1,2,*,† 1 Epidemiology of Highly Pathogenic Microorganisms, Robert Koch Institute, 13353 Berlin, Germany; [email protected] (J.F.G.); [email protected] (M.U.); [email protected] (T.L.) 2 Viral Evolution, Robert Koch Institute, 13353 Berlin, Germany 3 Center for Infection and Immunity, Mailman School of Public Health, Columbia University, New York, NY 10027, USA; [email protected] (N.B.); [email protected] (J.G.); [email protected] (K.J.); [email protected] (B.L.); [email protected] (A.O.); [email protected] (N.M.) 4 Institute of Microbiology and Epizootics, Freie Universität Berlin, 14163 Berlin, Germany 5 World Wide Fund for Nature, Dzanga-Sangha Protected Areas, Bangui BP 1053, Central African Republic; [email protected] 6 Laboratoire National D’appui au Développement Agricole/Laboratoire Central de Pathologie Animale, Bingerville BP206, Côte d’Ivoire; [email protected] * Correspondence: [email protected] (T.B.); [email protected] (S.C.-S.); [email protected] (W.I.L.); [email protected] (F.H.L.) † These authors contributed equally to the manuscript. Received: 28 February 2019; Accepted: 16 March 2019; Published: 19 March 2019 Abstract: New technologies enable viral discovery in a diversity of hosts, providing insights into viral evolution.