Full Text (PDF)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 Are Associated with Hearing Loss in Human and Mouse
RESEARCH ARTICLE Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in human and mouse Rizwan Yousaf1, Chunfang Gu2, Zubair M. Ahmed3, Shaheen N. Khan4, Thomas B. Friedman5, Sheikh Riazuddin6, Stephen B. Shears2, Saima Riazuddin1* a1111111111 1 Laboratory of Molecular Genetics, Department of Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United States of America, 2 Signal Transduction Laboratory, a1111111111 National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, a1111111111 NC, United States of America, 3 Laboratory of Neurogenetics and Regenerative Medicine, Department of a1111111111 Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United a1111111111 States of America, 4 National Center for Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan, 5 Section on Human Genetics, Laboratory of Molecular Genetics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD, United States of America, 6 Shaheed Zulfiqar Ali Bhutto Medical University, Pakistan Institute of Medical Sciences, Islamabad, Pakistan * [email protected] OPEN ACCESS Citation: Yousaf R, Gu C, Ahmed ZM, Khan SN, Friedman TB, Riazuddin S, et al. (2018) Mutations Abstract in Diphosphoinositol-Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in Autosomal recessive nonsyndromic hearing loss is a genetically heterogeneous disorder. human and mouse. PLoS Genet 14(3): e1007297. https://doi.org/10.1371/journal.pgen.1007297 Here, we report a severe-to-profound sensorineural hearing loss locus, DFNB100 on chro- mosome 5q13.2-q23.2. Exome enrichment followed by massive parallel sequencing Editor: Karen B. Avraham, Tel Aviv University, ISRAEL revealed a c.2510G>A transition variant in PPIP5K2 that segregated with DFNB100-associ- ated hearing loss in two large apparently unrelated Pakistani families. -

Transcriptome Analyses of Rhesus Monkey Pre-Implantation Embryos Reveal A
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Transcriptome analyses of rhesus monkey pre-implantation embryos reveal a reduced capacity for DNA double strand break (DSB) repair in primate oocytes and early embryos Xinyi Wang 1,3,4,5*, Denghui Liu 2,4*, Dajian He 1,3,4,5, Shengbao Suo 2,4, Xian Xia 2,4, Xiechao He1,3,6, Jing-Dong J. Han2#, Ping Zheng1,3,6# Running title: reduced DNA DSB repair in monkey early embryos Affiliations: 1 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 2 Key Laboratory of Computational Biology, CAS Center for Excellence in Molecular Cell Science, Collaborative Innovation Center for Genetics and Developmental Biology, Chinese Academy of Sciences-Max Planck Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China 3 Yunnan Key Laboratory of Animal Reproduction, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 4 University of Chinese Academy of Sciences, Beijing, China 5 Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650204, China 6 Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China * Xinyi Wang and Denghui Liu contributed equally to this work 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press # Correspondence: Jing-Dong J. Han, Email: [email protected]; Ping Zheng, Email: [email protected] Key words: rhesus monkey, pre-implantation embryo, DNA damage 2 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Pre-implantation embryogenesis encompasses several critical events including genome reprogramming, zygotic genome activation (ZGA) and cell fate commitment. -

A 1.37-Mb 12P11.22-P11.21 Deletion Coincident with a 367-Kb 22Q11.2
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Taiwanese Journal of Obstetrics & Gynecology 53 (2014) 74e78 Contents lists available at ScienceDirect Taiwanese Journal of Obstetrics & Gynecology journal homepage: www.tjog-online.com Short Communication A 1.37-Mb 12p11.22ep11.21 deletion coincident with a 367-kb 22q11.2 duplication detected by array comparative genomic hybridization in an adolescent girl with autism and difficulty in self-care of menstruation Chih-Ping Chen a,b,c,d,e,f,*, Shuan-Pei Lin b,g,h,i, Schu-Rern Chern b, Peih-Shan Wu j, Jun-Wei Su a,k, Chen-Chi Lee a, Wayseen Wang b,l a Department of Obstetrics and Gynecology, Mackay Memorial Hospital, Taipei, Taiwan b Department of Medical Research, Mackay Memorial Hospital, Taipei, Taiwan c Department of Biotechnology, Asia University, Taichung, Taiwan d School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan e Institute of Clinical and Community Health Nursing, National Yang-Ming University, Taipei, Taiwan f Department of Obstetrics and Gynecology, School of Medicine, National Yang-Ming University, Taipei, Taiwan g Department of Medicine, Mackay Medical College, New Taipei City, Taiwan h Department of Pediatrics, Mackay Memorial Hospital, Taipei, Taiwan i Mackay Junior College of Medicine, Nursing, and Management, Taipei, Taiwan j Gene Biodesign Co. Ltd, Taipei, Taiwan k Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan l Department of Bioengineering, Tatung University, Taipei, Taiwan article info abstract Article history: Objective: To present an array comparative genomic hybridization (aCGH) characterization of a 12p11.22 Accepted 21 October 2013 ep11.21 microdeletion and 22q11.2 microduplication in an adolescent girl with autism, mental retar- dation, facial dysmorphism, microcephaly, behavior problems, and an apparently balanced reciprocal Keywords: translocation of t(8;12)(q24.3;p11.2). -

Evidence for Differential Alternative Splicing in Blood of Young Boys With
Stamova et al. Molecular Autism 2013, 4:30 http://www.molecularautism.com/content/4/1/30 RESEARCH Open Access Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders Boryana S Stamova1,2,5*, Yingfang Tian1,2,4, Christine W Nordahl1,3, Mark D Shen1,3, Sally Rogers1,3, David G Amaral1,3 and Frank R Sharp1,2 Abstract Background: Since RNA expression differences have been reported in autism spectrum disorder (ASD) for blood and brain, and differential alternative splicing (DAS) has been reported in ASD brains, we determined if there was DAS in blood mRNA of ASD subjects compared to typically developing (TD) controls, as well as in ASD subgroups related to cerebral volume. Methods: RNA from blood was processed on whole genome exon arrays for 2-4–year-old ASD and TD boys. An ANCOVA with age and batch as covariates was used to predict DAS for ALL ASD (n=30), ASD with normal total cerebral volumes (NTCV), and ASD with large total cerebral volumes (LTCV) compared to TD controls (n=20). Results: A total of 53 genes were predicted to have DAS for ALL ASD versus TD, 169 genes for ASD_NTCV versus TD, 1 gene for ASD_LTCV versus TD, and 27 genes for ASD_LTCV versus ASD_NTCV. These differences were significant at P <0.05 after false discovery rate corrections for multiple comparisons (FDR <5% false positives). A number of the genes predicted to have DAS in ASD are known to regulate DAS (SFPQ, SRPK1, SRSF11, SRSF2IP, FUS, LSM14A). In addition, a number of genes with predicted DAS are involved in pathways implicated in previous ASD studies, such as ROS monocyte/macrophage, Natural Killer Cell, mTOR, and NGF signaling. -

DNA Replication Stress Response Involving PLK1, CDC6, POLQ
DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients C. Allera-Moreau, I. Rouquette, B. Lepage, N. Oumouhou, M. Walschaerts, E. Leconte, V. Schilling, K. Gordien, L. Brouchet, Mb Delisle, et al. To cite this version: C. Allera-Moreau, I. Rouquette, B. Lepage, N. Oumouhou, M. Walschaerts, et al.. DNA replica- tion stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis, Nature Publishing Group: Open Access Journals - Option C, 2012, 1, pp.e30. 10.1038/oncsis.2012.29. hal-00817701 HAL Id: hal-00817701 https://hal.archives-ouvertes.fr/hal-00817701 Submitted on 9 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License Citation: Oncogenesis (2012) 1, e30; doi:10.1038/oncsis.2012.29 & 2012 Macmillan Publishers Limited All rights reserved 2157-9024/12 www.nature.com/oncsis ORIGINAL ARTICLE DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients C Allera-Moreau1,2,7, I Rouquette2,7, B Lepage3, N Oumouhou3, M Walschaerts4, E Leconte5, V Schilling1, K Gordien2, L Brouchet2, MB Delisle1,2, J Mazieres1,2, JS Hoffmann1, P Pasero6 and C Cazaux1 Lung cancer is the leading cause of cancer deaths worldwide. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

A CRISPR-Based Screen for Hedgehog Signaling Provides Insights Into Ciliary Function and Ciliopathies
ARTICLES https://doi.org/10.1038/s41588-018-0054-7 A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies David K. Breslow 1,2,7*, Sascha Hoogendoorn 3,7, Adam R. Kopp2, David W. Morgens4, Brandon K. Vu2, Margaret C. Kennedy1, Kyuho Han4, Amy Li4, Gaelen T. Hess4, Michael C. Bassik4, James K. Chen 3,5* and Maxence V. Nachury 2,6* Primary cilia organize Hedgehog signaling and shape embryonic development, and their dysregulation is the unifying cause of ciliopathies. We conducted a functional genomic screen for Hedgehog signaling by engineering antibiotic-based selection of Hedgehog-responsive cells and applying genome-wide CRISPR-mediated gene disruption. The screen can robustly identify factors required for ciliary signaling with few false positives or false negatives. Characterization of hit genes uncovered novel components of several ciliary structures, including a protein complex that contains δ -tubulin and ε -tubulin and is required for centriole maintenance. The screen also provides an unbiased tool for classifying ciliopathies and showed that many congenital heart disorders are caused by loss of ciliary signaling. Collectively, our study enables a systematic analysis of ciliary function and of ciliopathies, and also defines a versatile platform for dissecting signaling pathways through CRISPR-based screening. he primary cilium is a surface-exposed microtubule-based approach. Indeed, most studies to date have searched for genes that compartment that serves as an organizing center for diverse either intrinsically affect cell growth or affect sensitivity to applied Tsignaling pathways1–3. Mutations affecting cilia cause ciliopa- perturbations16–23. thies, a group of developmental disorders including Joubert syn- Here, we engineered a Hh-pathway-sensitive reporter to enable drome, Meckel syndrome (MKS), nephronophthisis (NPHP), and an antibiotic-based selection platform. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Prioritization of Metabolic Genes As Novel Therapeutic Targets in Estrogen-Receptor Negative Breast Tumors Using Multi-Omics Data and Text Mining
www.oncotarget.com Oncotarget, 2019, Vol. 10, (No. 39), pp: 3894-3909 Research Paper Prioritization of metabolic genes as novel therapeutic targets in estrogen-receptor negative breast tumors using multi-omics data and text mining Dinesh Kumar Barupal1,*, Bei Gao1,*, Jan Budczies2, Brett S. Phinney4, Bertrand Perroud4, Carsten Denkert2,3 and Oliver Fiehn1 1West Coast Metabolomics Center, University of California, Davis, CA, USA 2Institute of Pathology, Charité University Hospital, Berlin, Germany 3German Institute of Pathology, Philipps-University Marburg, Marburg, Germany 4UC Davis Genome Center, University of California, Davis, CA, USA *Co-first authors and contributed equally to this work Correspondence to: Oliver Fiehn, email: [email protected] Keywords: set-enrichment; ChemRICH; multi-omics; metabolic networks; candidate gene prioritization Received: March 12, 2019 Accepted: May 13, 2019 Published: June 11, 2019 Copyright: Barupal et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Estrogen-receptor negative (ERneg) breast cancer is an aggressive breast cancer subtype in the need for new therapeutic options. We have analyzed metabolomics, proteomics and transcriptomics data for a cohort of 276 breast tumors (MetaCancer study) and nine public transcriptomics datasets using univariate statistics, meta- analysis, Reactome pathway analysis, biochemical network mapping and text mining of metabolic genes. In the MetaCancer cohort, a total of 29% metabolites, 21% proteins and 33% transcripts were significantly different (raw p <0.05) between ERneg and ERpos breast tumors. -
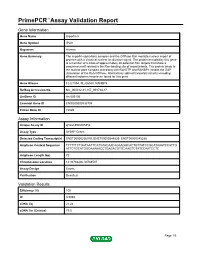
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name importin 8 Gene Symbol IPO8 Organism Human Gene Summary The importin-alpha/beta complex and the GTPase Ran mediate nuclear import of proteins with a classical nuclear localization signal. The protein encoded by this gene is a member of a class of approximately 20 potential Ran targets that share a sequence motif related to the Ran-binding site of importin-beta. This protein binds to the nuclear pore complex and along with RanGTP and RANBP1 inhibits the GAP stimulation of the Ran GTPase. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases FLJ21954, FLJ26580, RANBP8 RefSeq Accession No. NC_000012.11, NT_009714.17 UniGene ID Hs.505136 Ensembl Gene ID ENSG00000133704 Entrez Gene ID 10526 Assay Information Unique Assay ID qHsaCED0005354 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000256079, ENST00000544829, ENST00000545286 Amplicon Context Sequence TTTTTTCTGATAATTCATGTACAACAGAAGGCACTGTTATCCGCATAAATCCATTG ATTCTGCATGGCAAAAACCTGAGACGTGCAAGTCTATCCAATCCTC Amplicon Length (bp) 72 Chromosome Location 12:30783406-30783507 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9998 cDNA Cq 21.24 cDNA Tm (Celsius) 79.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 24.52 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report IPO8, Human Amplification Plot Amplification of cDNA generated from -

Mouse Ipo8 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ipo8 Knockout Project (CRISPR/Cas9) Objective: To create a Ipo8 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ipo8 gene (NCBI Reference Sequence: NM_001081113 ; Ensembl: ENSMUSG00000040029 ) is located on Mouse chromosome 6. 25 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 25 (Transcript: ENSMUST00000048418). Exon 2~4 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 2.81% of the coding region. Exon 2~4 covers 13.14% of the coding region. The size of effective KO region: ~6522 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 25 Legends Exon of mouse Ipo8 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 1322 bp section downstream of Exon 4 is aligned with itself to determine if there are tandem repeats.