Combining Landscape Genomics and Ecological Modelling To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Knowledge Status and Sampling Strategies to Maximize Cost-Benefit
www.nature.com/scientificreports OPEN Knowledge status and sampling strategies to maximize cost- beneft ratio of studies in landscape genomics of wild plants Alesandro Souza Santos* & Fernanda Amato Gaiotto To avoid local extinction due to the changes in their natural ecosystems, introduced by anthropogenic activities, species undergo local adaptation. Landscape genomics approach, through genome– environment association studies, has helped evaluate the local adaptation in natural populations. Landscape genomics, is still a developing discipline, requiring refnement of guidelines in sampling design, especially for studies conducted in the backdrop of stark socioeconomic realities of the rainforest ecologies, which are global biodiversity hotspots. In this study we aimed to devise strategies to improve the cost-beneft ratio of landscape genomics studies by surveying sampling designs and genome sequencing strategies used in existing studies. We conducted meta-analyses to evaluate the importance of sampling designs, in terms of (i) number of populations sampled, (ii) number of individuals sampled per population, (iii) total number of individuals sampled, and (iv) number of SNPs used in diferent studies, in discerning the molecular mechanisms underlying local adaptation of wild plant species. Using the linear mixed efects model, we demonstrated that the total number of individuals sampled and the number of SNPs used, signifcantly infuenced the detection of loci underlying the local adaptation. Thus, based on our fndings, in order to optimize the cost-beneft ratio of landscape genomics studies, we suggest focusing on increasing the total number of individuals sampled and using a targeted (e.g. sequencing capture) Pool-Seq approach and/or a random (e.g. RAD- Seq) Pool-Seq approach to detect SNPs and identify SNPs under selection for a given environmental cline. -

The Climatic and Genetic Heritage of Italian Goat Breeds with Genomic
www.nature.com/scientificreports OPEN The climatic and genetic heritage of Italian goat breeds with genomic SNP data Matteo Cortellari 1,16, Mario Barbato2,16, Andrea Talenti1,3*, Arianna Bionda1, Antonello Carta4, Roberta Ciampolini5, Elena Ciani6, Alessandra Crisà7, Stefano Frattini1, Emiliano Lasagna8, Donata Marletta9, Salvatore Mastrangelo10, Alessio Negro1, Ettore Randi11, Francesca M. Sarti8, Stefano Sartore12, Dominga Soglia12, Luigi Liotta13, Alessandra Stella14, Paolo Ajmone‑Marsan2, Fabio Pilla15, Licia Colli2 & Paola Crepaldi1 Local adaptation of animals to the environment can abruptly become a burden when faced with rapid climatic changes such as those foreseen for the Italian peninsula over the next 70 years. Our study investigates the genetic structure of the Italian goat populations and links it with the environment and how genetics might evolve over the next 50 years. We used one of the largest national datasets including > 1000 goats from 33 populations across the Italian peninsula collected by the Italian Goat Consortium and genotyped with over 50 k markers. Our results showed that Italian goats can be discriminated in three groups refective of the Italian geography and its geo‑political situation preceding the country unifcation around two centuries ago. We leveraged the remarkable genetic and geographical diversity of the Italian goat populations and performed landscape genomics analysis to disentangle the relationship between genotype and environment, fnding 64 SNPs intercepting genomic regions linked to growth, circadian rhythm, fertility, and infammatory response. Lastly, we calculated the hypothetical future genotypic frequencies of the most relevant SNPs identifed through landscape genomics to evaluate their long‑term efect on the genetic structure of the Italian goat populations. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Aberrant Sialylation in Cancer: Biomarker and Potential Target for Therapeutic Intervention?
cancers Review Aberrant Sialylation in Cancer: Biomarker and Potential Target for Therapeutic Intervention? Silvia Pietrobono * and Barbara Stecca * Tumor Cell Biology Unit, Core Research Laboratory, Institute for Cancer Research and Prevention (ISPRO), Viale Pieraccini 6, 50139 Florence, Italy * Correspondence: [email protected] (S.P.); [email protected] (B.S.); Tel.: +39-055-7944568 (S.P.); +39-055-7944567 (B.S.) Simple Summary: Sialylation is a post-translational modification that consists in the addition of sialic acid to growing glycan chains on glycoproteins and glycolipids. Aberrant sialylation is an established hallmark of several types of cancer, including breast, ovarian, pancreatic, prostate, colorectal and lung cancers, melanoma and hepatocellular carcinoma. Hypersialylation can be the effect of increased activity of sialyltransferases and results in an excess of negatively charged sialic acid on the surface of cancer cells. Sialic acid accumulation contributes to tumor progression by several paths, including stimulation of tumor invasion and migration, and enhancing immune evasion and tumor cell survival. In this review we explore the mechanisms by which sialyltransferases promote cancer progression. In addition, we provide insights into the possible use of sialyltransferases as biomarkers for cancer and summarize findings on the development of sialyltransferase inhibitors as potential anti-cancer treatments. Abstract: Sialylation is an integral part of cellular function, governing many biological processes Citation: Pietrobono, S.; Stecca, B. including cellular recognition, adhesion, molecular trafficking, signal transduction and endocytosis. Aberrant Sialylation in Cancer: Sialylation is controlled by the levels and the activities of sialyltransferases on glycoproteins and Biomarker and Potential Target for lipids. Altered gene expression of these enzymes in cancer yields to cancer-specific alterations of Therapeutic Intervention? Cancers glycoprotein sialylation. -

Title: the Genomic Consequences of Hybridization Authors
Title: The genomic consequences of hybridization Authors: Benjamin M Moran1,2*+, Cheyenne Payne1,2*+, Quinn Langdon1, Daniel L Powell1,2, Yaniv Brandvain3, Molly Schumer1,2,4+ Affiliations: 1Department of Biology, Stanford University, Stanford, CA, USA 2Centro de Investigaciones Científicas de las Huastecas “Aguazarca”, A.C., Calnali, Hidalgo, Mexico 3Department of Ecology, Evolution & Behavior and Plant and Microbial Biology, University of Minnesota, St. Paul, MN, USA 4Hanna H. Gray Fellow, Howard Hughes Medical Institute, Stanford, CA, USA *Contributed equally to this work +Correspondence: [email protected], [email protected], [email protected] Abstract In the past decade, advances in genome sequencing have allowed researchers to uncover the history of hybridization in diverse groups of species, including our own. Although the field has made impressive progress in documenting the extent of natural hybridization, both historical and recent, there are still many unanswered questions about its genetic and evolutionary consequences. Recent work has suggested that the outcomes of hybridization in the genome may be in part predictable, but many open questions about the nature of selection on hybrids and the biological variables that shape such selection have hampered progress in this area. We discuss what is known about the mechanisms that drive changes in ancestry in the genome after hybridization, highlight major unresolved questions, and discuss their implications for the predictability of genome evolution after hybridization. Introduction Recent evidence has shown that hybridization between species is common. Hybridization is widespread across the tree of life, spanning both ancient and recent timescales and a broad range of divergence levels between taxa [1–10]. This appreciation of the prevalence of hybridization has renewed interest among researchers in understanding its consequences. -

Current Status, Future Opportunities, and Remaining Challenges in Landscape Genetics
Chapter 14 CURRENT STATUS, FUTURE OPPORTUNITIES, AND REMAINING CHALLENGES IN LANDSCAPE GENETICS 1 2 3 4 Niko Balkenhol, Samuel A. Cushman, Lisette P. Waits, and Andrew Storfer 1 Department of Wildlife Sciences, University of Göttingen, Germany 2 Forest and Woodlands Ecosystems Program, Rocky Mountain Research Station, United States Forest Service, USA 3 Fish and Wildlife Sciences, University of Idaho, USA 4 School of Biological Sciences, Washington State University, USA 14.1 INTRODUCTION essentially replaced the isolation-by-distance paradigm with spatially explicit tests of effects of landscape varia- Just over a decade ago, advances in geographic informa- bles on genetic population structure. More generally, tion systems and genetic methodology helped usher in landscape genetics has advanced the field of evolution- the field of landscape genetics, an amalgamation of ary ecology by providing a direct focus on relationships landscape ecology, population genetics, and spatial sta- between landscape patterns and population processes, tistics aimed at testing how landscape heterogeneity such as gene flow, selection, and genetic drift. shapes patterns of spatial genetic variation. It is clear Since the inception of landscape genetics, there have that the tools developed in landscape genetics have been many calls for: (1) better communication among shaped a new paradigm for conducting empirical popu- spatial statisticians, landscape ecologists, and population lation genetics studies; although population genetics geneticists (Storfer et al. 2007; Balkenhol et al. 2009a); theory itself remains largely unchanged, the majority (2) more rigorous hypothesis testing (Storfer et al. 2007, of empirical studies of spatial genetic structure now 2010; Cushman & Landguth 2010; Segelbacher et al. include spatially explicit tests of landscape influences 2010; Manel & Holderegger 2013); (3) increased con- on gene flow. -

Downloaded 18 July 2014 with a 1% False Discovery Rate (FDR)
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer Permalink https://escholarship.org/uc/item/0t47b9ws Author Spiciarich, David Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer by David Spiciarich A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Carolyn R. Bertozzi, Co-Chair Professor David E. Wemmer, Co-Chair Professor Matthew B. Francis Professor Amy E. Herr Fall 2017 Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer © 2017 by David Spiciarich Abstract Chemical glycoproteomics for identification and discovery of glycoprotein alterations in human cancer by David Spiciarich Doctor of Philosophy in Chemistry University of California, Berkeley Professor Carolyn R. Bertozzi, Co-Chair Professor David E. Wemmer, Co-Chair Changes in glycosylation have long been appreciated to be part of the cancer phenotype; sialylated glycans are found at elevated levels on many types of cancer and have been implicated in disease progression. However, the specific glycoproteins that contribute to cell surface sialylation are not well characterized, specifically in bona fide human cancer. Metabolic and bioorthogonal labeling methods have previously enabled enrichment and identification of sialoglycoproteins from cultured cells and model organisms. The goal of this work was to develop technologies that can be used for detecting changes in glycoproteins in clinical models of human cancer. -
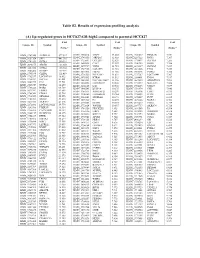
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -

Recent Advances in Conservation and Population Genomics Data Analysis
Received: 20 February 2018 | Revised: 8 May 2018 | Accepted: 21 May 2018 DOI: 10.1111/eva.12659 MEETING REPORT Recent advances in conservation and population genomics data analysis Sarah Hendricks1 | Eric C. Anderson2,3 | Tiago Antao4 | Louis Bernatchez5 | Brenna R. Forester6 | Brittany Garner7,8 | Brian K. Hand7 | Paul A. Hohenlohe1 | Martin Kardos7 | Ben Koop9 | Arun Sethuraman10 | Robin S. Waples11 | Gordon Luikart7,8 1Institute for Bioinformatics and Evolutionary Studies, University of Idaho, Moscow, Idaho 2Fisheries Ecology Division, Southwest Fisheries Science Center, National Marine Fisheries Service, National Oceanic and Atmospheric Administration, Santa Cruz, California 3University of California, Santa Cruz, California 4Division of Biological Sciences, University of Montana, Missoula, Montana 5Département de Biologie, Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Québec, Québec, Canada 6Department of Biology, Colorado State University, Fort Collins, Colorado 7Flathead Lake Biological Station, Montana Conservation Genomics Laboratory, Division of Biological Science, University of Montana, Missoula, Montana 8Wildlife Program, Fish and Wildlife Genomics Group, College of Forestry and Conservation, University of Montana, Missoula, Montana 9Department of Biology, Centre for Biomedical Research, University of Victoria, Victoria, British Columbia, Canada 10Department of Biological Sciences, California State University San Marcos, San Marcos, California 11NOAA Fisheries, Northwest Fisheries Science Center, -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Upregulation of Mir-181C Inhibits Chemoresistance by Targeting ST8SIA4 in Chronic Myelocytic Leukemia
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 37 Research Paper Upregulation of miR-181c inhibits chemoresistance by targeting ST8SIA4 in chronic myelocytic leukemia Lifen Zhao1, Yan Li2, Xiaobo Song3, Huimin Zhou4, Nana Li1, Yuan Miao1, Li Jia1 1College of Laboratory Medicine, Dalian Medical University, Dalian, Liaoning Province, China 2Department of Clinical Laboratory, the First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning Province, China 3Department of Medical Biology, Faculty of Health Sciences, University of Tromsø, Tromsø, Norway 4Department of Microbiology, Dalian Medical University, Dalian, Liaoning Province, China Correspondence to: Li Jia, email: [email protected] Keywords: miR-181c, ST8SIA4, chronic myelocytic leukemia, PI3K/AKT, chemoresistance Received: April 04, 2016 Accepted: July 10, 2016 Published: August 04, 2016 ABSTRACT Chemotherapy resistance frequently drives tumor progression. Increased expression of ST8SIA4 has been reported in diverse carcinomas and highly correlates with leukemia multidrug resistance (MDR). MicroRNAs (miRNA) are widely recognized as key players in cancer progression and drug resistance. Here, to explore whether miRNA modulates the sensitivity of chronic myelocytic leukemia (CML) to chemotherapeutic agents and regulates ST8SIA4 expression, we analyzed the complete miRNA expression profile and found a subset of miRNAs specifically dysregulated in adriamycin-resistant CML cell line K562/ADR and its parent cell line K562. Compared with three pairs of CML cell lines and 38 clinical samples of peripheral blood mononuclear cells (PBMC) of CML patients, miR-181c expression was down- regulated in drug-resistant cell lines and CML/MDR samples. Altered expression levels of miR-181c influenced the MDR phenotypes of K562 and K562/ADR. Reporter-gene assay showed that miR-181c directly targeted and inhibited the ST8SIA4 expression, as well as miR-181c was inversely correlated with the levels of ST8SIA4 expression in CML cell lines and samples.