Gene Evolution and Gene Expression After Whole Genome Duplication in Fish
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ecology and Conservation of Mudminnow Species Worldwide
FEATURE Ecology and Conservation of Mudminnow Species Worldwide Lauren M. Kuehne Ecología y conservación a nivel mundial School of Aquatic and Fishery Sciences, University of Washington, Seattle, WA 98195 de los lucios RESUMEN: en este trabajo, se revisa y resume la ecología Julian D. Olden y estado de conservación del grupo de peces comúnmente School of Aquatic and Fishery Sciences, University of Washington, Box conocido como “lucios” (anteriormente conocidos como 355020, Seattle, WA 98195, and Australian Rivers Institute, Griffith Uni- la familia Umbridae, pero recientemente reclasificados en versity, QLD, 4111, Australia. E-mail: [email protected] la Esocidae) los cuales se constituyen de sólo cinco espe- cies distribuidas en tres continentes. Estos peces de cuerpo ABSTRACT: We review and summarize the ecology and con- pequeño —que viven en hábitats de agua dulce y presentan servation status of the group of fishes commonly known as movilidad limitada— suelen presentar poblaciones aisla- “mudminnows” (formerly known as the family Umbridae but das a lo largo de distintos paisajes y son sujetos a las típi- recently reclassified as Esocidae), consisting of only five species cas amenazas que enfrentan las especies endémicas que distributed on three continents. These small-bodied fish—resid- se encuentran en contacto directo con los impactos antro- ing in freshwater habitats and exhibiting limited mobility—often pogénicos como la contaminación, alteración de hábitat occur in isolated populations across landscapes and are subject e introducción de especies no nativas. Aquí se resume el to conservation threats common to highly endemic species in conocimiento actual acerca de la distribución, relaciones close contact with anthropogenic impacts, such as pollution, filogenéticas, ecología y estado de conservación de cada habitat alteration, and nonnative species introductions. -

SR 53(5) 38-40.Pdf
M. GOSWAMI & ANIRBAN ROY RTICLE A EATURE F An understanding of the evolution of the electric organ from muscle cells in electric fi shes can open a new horizon in synthetic biology. Muscles in other vertebrates or invertebrates may be manipulated for generating electrical power in human organs such as heart, brain, and spinal cord. Since the last few decades, the the resting state, the internal potential development and working of electric amounts to -70mV to -80mV (depending organs inside the fi sh’s body has been upon the type of cell). This is termed as a sublime topic of interest for many resting potential or Nernst potential. The researchers. The scientifi c world is of negative sign in the membrane potential the opinion that the electric organs from signifi es the presence of the non-diffusible which electric discharges are produced anions and unequal distribution of ions have evolved half a dozen times in the across cytosol. HILE we humans have to generate environment. Variations of ionic concentration electricity to take care of many W inside and outside the cell as well as activities, there are fi shes that produce difference in the permeability of cell their own electricity. Electric fi shes and Bioelectricity membrane to diverse ions are responsible A fi sh capable of generating electric fi elds Within the aquatic world, there for the existence of resting potential. is said to be electrogenic while a fi sh are hundreds of electric fi shes. Charles Usually K+, Na+, Cl-, Ca2+ ions are that can detect electric fi elds is said to be Darwin had recognised electric fi shes as widely available in the intracellular and electroreceptive. -

Convergent Evolution of Mechanically Optimal Locomotion in Aquatic Invertebrates and Vertebrates
RESEARCH ARTICLE Convergent Evolution of Mechanically Optimal Locomotion in Aquatic Invertebrates and Vertebrates Rahul Bale1, Izaak D. Neveln2, Amneet Pal Singh Bhalla1, Malcolm A. MacIver1,2,3☯*, Neelesh A. Patankar1☯* 1 Department of Mechanical Engineering, Northwestern University, Evanston, Illinois, United States of America, 2 Department of Biomedical Engineering, Northwestern University, Evanston, Illinois, United States of America, 3 Department of Neurobiology, Northwestern University, Evanston, Illinois, United States of America ☯ These authors contributed equally to this work. * [email protected] (NAP); [email protected] (MAM) Abstract OPEN ACCESS Examples of animals evolving similar traits despite the absence of that trait in the last com- Citation: Bale R, Neveln ID, Bhalla APS, MacIver MA, Patankar NA (2015) Convergent Evolution of mon ancestor, such as the wing and camera-type lens eye in vertebrates and invertebrates, Mechanically Optimal Locomotion in Aquatic are called cases of convergent evolution. Instances of convergent evolution of locomotory Invertebrates and Vertebrates. PLoS Biol 13(4): patterns that quantitatively agree with the mechanically optimal solution are very rare. Here, e1002123. doi:10.1371/journal.pbio.1002123 we show that, with respect to a very diverse group of aquatic animals, a mechanically opti- Academic Editor: Anders Hedenström, Lund mal method of swimming with elongated fins has evolved independently at least eight times University, SWEDEN in both vertebrate and invertebrate swimmers across three different phyla. Specifically, if we Received: September 29, 2014 take the length of an undulation along an animal’s fin during swimming and divide it by the Accepted: March 6, 2015 mean amplitude of undulations along the fin length, the result is consistently around twenty. -

SPECIAL PUBLICATION 6 the Effects of Marine Debris Caused by the Great Japan Tsunami of 2011
PICES SPECIAL PUBLICATION 6 The Effects of Marine Debris Caused by the Great Japan Tsunami of 2011 Editors: Cathryn Clarke Murray, Thomas W. Therriault, Hideaki Maki, and Nancy Wallace Authors: Stephen Ambagis, Rebecca Barnard, Alexander Bychkov, Deborah A. Carlton, James T. Carlton, Miguel Castrence, Andrew Chang, John W. Chapman, Anne Chung, Kristine Davidson, Ruth DiMaria, Jonathan B. Geller, Reva Gillman, Jan Hafner, Gayle I. Hansen, Takeaki Hanyuda, Stacey Havard, Hirofumi Hinata, Vanessa Hodes, Atsuhiko Isobe, Shin’ichiro Kako, Masafumi Kamachi, Tomoya Kataoka, Hisatsugu Kato, Hiroshi Kawai, Erica Keppel, Kristen Larson, Lauran Liggan, Sandra Lindstrom, Sherry Lippiatt, Katrina Lohan, Amy MacFadyen, Hideaki Maki, Michelle Marraffini, Nikolai Maximenko, Megan I. McCuller, Amber Meadows, Jessica A. Miller, Kirsten Moy, Cathryn Clarke Murray, Brian Neilson, Jocelyn C. Nelson, Katherine Newcomer, Michio Otani, Gregory M. Ruiz, Danielle Scriven, Brian P. Steves, Thomas W. Therriault, Brianna Tracy, Nancy C. Treneman, Nancy Wallace, and Taichi Yonezawa. Technical Editor: Rosalie Rutka Please cite this publication as: The views expressed in this volume are those of the participating scientists. Contributions were edited for Clarke Murray, C., Therriault, T.W., Maki, H., and Wallace, N. brevity, relevance, language, and style and any errors that [Eds.] 2019. The Effects of Marine Debris Caused by the were introduced were done so inadvertently. Great Japan Tsunami of 2011, PICES Special Publication 6, 278 pp. Published by: Project Designer: North Pacific Marine Science Organization (PICES) Lori Waters, Waters Biomedical Communications c/o Institute of Ocean Sciences Victoria, BC, Canada P.O. Box 6000, Sidney, BC, Canada V8L 4B2 Feedback: www.pices.int Comments on this volume are welcome and can be sent This publication is based on a report submitted to the via email to: [email protected] Ministry of the Environment, Government of Japan, in June 2017. -

ILLEGAL FISHING Which Fish Species Are at Highest Risk from Illegal and Unreported Fishing?
ILLEGAL FISHING Which fish species are at highest risk from illegal and unreported fishing? October 2015 CONTENTS EXECUTIVE SUMMARY 3 INTRODUCTION 4 METHODOLOGY 5 OVERALL FINDINGS 9 NOTES ON ESTIMATES OF IUU FISHING 13 Tunas 13 Sharks 14 The Mediterranean 14 US Imports 15 CONCLUSION 16 CITATIONS 17 OCEAN BASIN PROFILES APPENDIX 1: IUU Estimates for Species Groups and Ocean Regions APPENDIX 2: Estimates of IUU Risk for FAO Assessed Stocks APPENDIX 3: FAO Ocean Area Boundary Descriptions APPENDIX 4: 2014 U.S. Edible Imports of Wild-Caught Products APPENDIX 5: Overexploited Stocks Categorized as High Risk – U.S. Imported Products Possibly Derived from Stocks EXECUTIVE SUMMARY New analysis by World Wildlife Fund (WWF) finds that over 85 percent of global fish stocks can be considered at significant risk of Illegal, Unreported, and Unregulated (IUU) fishing. This evaluation is based on the most recent comprehensive estimates of IUU fishing and includes the worlds’ major commercial stocks or species groups, such as all those that are regularly assessed by the United Nations Food and Agriculture Organization (FAO). Based on WWF’s findings, the majority of the stocks, 54 percent, are categorized as at high risk of IUU, with an additional 32 perent judged to be at moderate risk. Of the 567 stocks that were assessed, the findings show that 485 stocks fall into these two categories. More than half of the world’s most overexploited stocks are at the highest risk of IUU fishing. Examining IUU risk by location, the WWF analysis shows that in more than one-third of the world’s ocean basins as designated by the FAO, all of these stocks were at high or moderate risk of IUU fishing. -

Downloaded from NCBI Genbank (Benson Et Al
THE UNIVERSITY OF CHICAGO EVOLUTION IN FRESH WATERS DURING THE GREAT AMERICAN INTERCHANGE A DISSERTATION SUBMITTED TO THE FACULTY OF THE DIVISION OF THE BIOLOGICAL SCIENCES AND THE PRITZKER SCHOOL OF MEDICINE IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY COMMITTEE ON EVOLUTIONARY BIOLOGY BY TIMOTHY SOSA CHICAGO, ILLINOIS DECEMBER 2017 Table of Contents List of Tables . iii List of Figures . iv Acknowledgments . vi Chapter 1: Introduction . 1 Chapter 2: Broadly sampled phylogeny of Characiformes reveals repeated colonization of North America and paraphyly of Characiformes sensu stricto . 8 Chapter 3: No evidence for filtering of eco-morphology in characiform lineages during the Great American Interchange . 17 Chapter 4: Both elevation and species identity strongly predict body shape in Astyanax tetras . 27 Chapter 5: Diet may mediate potential range expansions of Neotropical fishes under climate change . 39 Chapter 6: Discussion . 52 References . 57 Appendix: List of specimens newly sequenced for this study . 67 ii List of Tables 1.1 Recognized families in the order Characiformes . 5 2.1 Fossil occurrences used for time-calibration . 11 4.1 Distances in morphospace among tetra populations . 32 5.1 Variables determining the range limits of Astyanax . 45 5.2 Variables determining the range limits of Brycon . 47 5.3 Variables determining the range limits of Roeboides . 49 iii List of Figures 1.1 Hypothetical relationships among ostariophysan groups . 4 2.1 Phylogeny of Characiformes as inferred from myh6 locus . 13 3.1 Landmark configuration for geometric morphometrics . 19 3.2 Morphospace occupation in North and South American characins . 21 3.3 Deformation grids showing axes of shape variation among characins . -

Intrinsic Vulnerability in the Global Fish Catch
The following appendix accompanies the article Intrinsic vulnerability in the global fish catch William W. L. Cheung1,*, Reg Watson1, Telmo Morato1,2, Tony J. Pitcher1, Daniel Pauly1 1Fisheries Centre, The University of British Columbia, Aquatic Ecosystems Research Laboratory (AERL), 2202 Main Mall, Vancouver, British Columbia V6T 1Z4, Canada 2Departamento de Oceanografia e Pescas, Universidade dos Açores, 9901-862 Horta, Portugal *Email: [email protected] Marine Ecology Progress Series 333:1–12 (2007) Appendix 1. Intrinsic vulnerability index of fish taxa represented in the global catch, based on the Sea Around Us database (www.seaaroundus.org) Taxonomic Intrinsic level Taxon Common name vulnerability Family Pristidae Sawfishes 88 Squatinidae Angel sharks 80 Anarhichadidae Wolffishes 78 Carcharhinidae Requiem sharks 77 Sphyrnidae Hammerhead, bonnethead, scoophead shark 77 Macrouridae Grenadiers or rattails 75 Rajidae Skates 72 Alepocephalidae Slickheads 71 Lophiidae Goosefishes 70 Torpedinidae Electric rays 68 Belonidae Needlefishes 67 Emmelichthyidae Rovers 66 Nototheniidae Cod icefishes 65 Ophidiidae Cusk-eels 65 Trachichthyidae Slimeheads 64 Channichthyidae Crocodile icefishes 63 Myliobatidae Eagle and manta rays 63 Squalidae Dogfish sharks 62 Congridae Conger and garden eels 60 Serranidae Sea basses: groupers and fairy basslets 60 Exocoetidae Flyingfishes 59 Malacanthidae Tilefishes 58 Scorpaenidae Scorpionfishes or rockfishes 58 Polynemidae Threadfins 56 Triakidae Houndsharks 56 Istiophoridae Billfishes 55 Petromyzontidae -
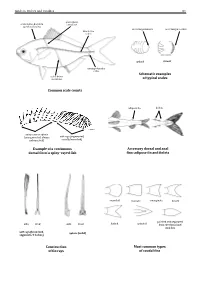
Field Identification Guide to the Living Marine Resources in Kenya
Guide to Orders and Families 81 lateral line scales above scales before dorsal fin outer margin smooth outer margin toothed (predorsal scales) lateral–line 114 scales cycloid ctenoidِّ scales circumpeduncular Schematic examples lateral line of typical scales scales below Common scale counts adipose fin finlets soft rays (segmented, spinyunbranched) rays or spines usually branched) (unsegmented, always Example of a continuous Accessory dorsal and anal dorsal fin of a spiny–rayed fish fins: adipose fin and finlets rounded truncate emarginate lunate side front side front from the dorsal and pointed and separated forked pointed soft rays (branched, spines (solid) segments, 2 halves) anal fins Construction Most common types of fin rays of caudal fins 82 Bony Fishes GUIDE TO ORDERS AND FAMILIES Order ELOPIFORMES – Tarpons and allies Fin spines absent; a single dorsal fin located above middle of body; pelvic fins in abdominal position; lateral line present; 23–25 branchiostegal rays; upper jaw extending past eye; tip of snout not overhanging mouth; colour silvery. ELOPIDAE Page 121 very small scales Ladyfishes To 90 cm. Coastal marine waters and estuaries; pelagic. A single species included in the Guide to Species.underside of head large mouth gular plate MEGALOPIDAE Page 121 last ray long Tarpons large scales To 55 cm. Coastal marine waters and estuaries; pelagic. A single species included in the Guide to Species.underside of head gular plate Order ALBULIFORMES – Bonefishes Fin spines absent; a single dorsal fin located above middle of body; pelvic fins in abdominal position; lateral line present; 6–16 branchiostegal rays; upper jaw not extending as far as front of eye; tip of snout overhanging mouth; colour silvery. -

Evolution of the Pgc-1 Protein Family in the Control of Oxidative Metabolism in Vertebrates
EVOLUTION OF THE PGC-1 PROTEIN FAMILY IN THE CONTROL OF OXIDATIVE METABOLISM IN VERTEBRATES by Christophe Marie Renaud Le Moine A thesis submitted to the Department of Biology In conformity with the requirements for the degree of Doctor of Philosophy Queen’s University Kingston, Ontario, Canada (July, 2008) Copyright © Christophe Marie Renaud Le Moine, 2008 Abstract Mitochondrial biogenesis requires an intricate transcriptional coordination between the nuclear and mitochondrial genomes to establish the structural and functional components of the organelle. This coordination is paramount in vertebrate muscles where oxidative capacity must be adjusted to meet varying energy demands. I investigated the regulatory circuits controlling mitochondrial content in vertebrate muscle in the context of development, adaptation to nutritional status and temperature, and in an evolutionary perspective. Initial experiments focused on the role of transcriptional regulators in the metabolic changes in the myocardium of aging rat. I hypothesized that the changes in oxidative capacity associated with aging would be primarily driven by the peroxisome proliferator activated-receptors (PPARs), the nuclear respiratory factors (NRFs) and their common coactivator PPAR coactivator-1 (PGC-1 . However, the reduction in oxidative capacity in the heart of old rats was independent of these regulatory axes and occurred partially through post-transcriptional processes. The next series of experiments investigated the transcriptional networks regulating the metabolic remodelling in goldfish subjected to dietary and temperature stress. As a potent regulator of mitochondrial proliferation in mammals, I hypothesized that PGC-1 assumed a similar role in lower vertebrates. Similar to their mammalian homologues, PPAR and NRF-1 assumed their respective roles in regulating lipid metabolism and mitochondrial proliferation in goldfish. -

Red List of Bangladesh 2015
Red List of Bangladesh Volume 1: Summary Chief National Technical Expert Mohammad Ali Reza Khan Technical Coordinator Mohammad Shahad Mahabub Chowdhury IUCN, International Union for Conservation of Nature Bangladesh Country Office 2015 i The designation of geographical entitles in this book and the presentation of the material, do not imply the expression of any opinion whatsoever on the part of IUCN, International Union for Conservation of Nature concerning the legal status of any country, territory, administration, or concerning the delimitation of its frontiers or boundaries. The biodiversity database and views expressed in this publication are not necessarily reflect those of IUCN, Bangladesh Forest Department and The World Bank. This publication has been made possible because of the funding received from The World Bank through Bangladesh Forest Department to implement the subproject entitled ‘Updating Species Red List of Bangladesh’ under the ‘Strengthening Regional Cooperation for Wildlife Protection (SRCWP)’ Project. Published by: IUCN Bangladesh Country Office Copyright: © 2015 Bangladesh Forest Department and IUCN, International Union for Conservation of Nature and Natural Resources Reproduction of this publication for educational or other non-commercial purposes is authorized without prior written permission from the copyright holders, provided the source is fully acknowledged. Reproduction of this publication for resale or other commercial purposes is prohibited without prior written permission of the copyright holders. Citation: Of this volume IUCN Bangladesh. 2015. Red List of Bangladesh Volume 1: Summary. IUCN, International Union for Conservation of Nature, Bangladesh Country Office, Dhaka, Bangladesh, pp. xvi+122. ISBN: 978-984-34-0733-7 Publication Assistant: Sheikh Asaduzzaman Design and Printed by: Progressive Printers Pvt. -

Volume 2E - Revised Baseline Ecological Risk Assessment Hudson River Pcbs Reassessment
PHASE 2 REPORT FURTHER SITE CHARACTERIZATION AND ANALYSIS VOLUME 2E - REVISED BASELINE ECOLOGICAL RISK ASSESSMENT HUDSON RIVER PCBS REASSESSMENT NOVEMBER 2000 For U.S. Environmental Protection Agency Region 2 and U.S. Army Corps of Engineers Kansas City District Book 2 of 2 Tables, Figures and Plates TAMS Consultants, Inc. Menzie-Cura & Associates, Inc. PHASE 2 REPORT FURTHER SITE CHARACTERIZATION AND ANALYSIS VOLUME 2E- REVISED BASELINE ECOLOGICAL RISK ASSESSMENT HUDSON RIVER PCBs REASSESSMENT RI/FS CONTENTS Volume 2E (Book 1 of 2) Page TABLE OF CONTENTS ........................................................ i LIST OF TABLES ........................................................... xiii LIST OF FIGURES ......................................................... xxv LIST OF PLATES .......................................................... xxvi EXECUTIVE SUMMARY ...................................................ES-1 1.0 INTRODUCTION .......................................................1 1.1 Purpose of Report .................................................1 1.2 Site History ......................................................2 1.2.1 Summary of PCB Sources to the Upper and Lower Hudson River ......4 1.2.2 Summary of Phase 2 Geochemical Analyses .......................5 1.2.3 Extent of Contamination in the Upper Hudson River ................5 1.2.3.1 PCBs in Sediment .....................................5 1.2.3.2 PCBs in the Water Column ..............................6 1.2.3.3 PCBs in Fish .........................................7 -

Foxr1 Is a Novel Maternal-Effect Gene in Fish That Is Required for Early Embryonic Success Caroline Cheung, Amélie Patinote, Yann Guiguen, Julien Bobe
Foxr1 is a novel maternal-effect gene in fish that is required for early embryonic success Caroline Cheung, Amélie Patinote, Yann Guiguen, Julien Bobe To cite this version: Caroline Cheung, Amélie Patinote, Yann Guiguen, Julien Bobe. Foxr1 is a novel maternal- effect gene in fish that is required for early embryonic success. PeerJ, PeerJ, 2018, 6,pp.1-20. 10.7717/peerj.5534. hal-01867990 HAL Id: hal-01867990 https://hal.archives-ouvertes.fr/hal-01867990 Submitted on 4 Sep 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License foxr1 is a novel maternal-effect gene in fish that is required for early embryonic success Caroline T. Cheung, Amélie Patinote, Yann Guiguen and Julien Bobe LPGP, UR1037 Fish Physiology and Genomics, INRA, Rennes, France ABSTRACT The family of forkhead box (Fox) transcription factors regulates gonadogenesis and embryogenesis, but the role of foxr1 in reproduction is unknown. Evolutionary history of foxr1 in vertebrates was examined and the gene was found to exist in most vertebrates, including mammals, ray-finned fish, amphibians, and sauropsids. By quantitative PCR and RNA-seq, we found that foxr1 had an ovarian-specific expression in zebrafish, a common feature of maternal-effect genes.