Immune Response and Endocytosis Pathways Are Associated with the Escape of Alzheimer’S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Podocyte Specific Knockdown of Klf15 in Podocin-Cre Klf15flox/Flox Mice Was Confirmed
SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1: Podocyte specific knockdown of Klf15 in Podocin-Cre Klf15flox/flox mice was confirmed. (A) Primary glomerular epithelial cells (PGECs) were isolated from 12-week old Podocin-Cre Klf15flox/flox and Podocin-Cre Klf15+/+ mice and cultured at 37°C for 1 week. Real-time PCR was performed for Nephrin, Podocin, Synaptopodin, and Wt1 mRNA expression (n=6, ***p<0.001, Mann-Whitney test). (B) Real- time PCR was performed for Klf15 mRNA expression (n=6, *p<0.05, Mann-Whitney test). (C) Protein was also extracted and western blot analysis for Klf15 was performed. The representative blot of three independent experiments is shown in the top panel. The bottom panel shows the quantification of Klf15 by densitometry (n=3, *p<0.05, Mann-Whitney test). (D) Immunofluorescence staining for Klf15 and Wt1 was performed in 12-week old Podocin-Cre Klf15flox/flox and Podocin-Cre Klf15+/+ mice. Representative images from four mice in each group are shown in the left panel (X 20). Arrows show colocalization of Klf15 and Wt1. Arrowheads show a lack of colocalization. Asterisk demonstrates nonspecific Wt1 staining. “R” represents autofluorescence from RBCs. In the right panel, a total of 30 glomeruli were selected in each mouse and quantification of Klf15 staining in the podocytes was determined by the ratio of Klf15+ and Wt1+ cells to Wt1+ cells (n=6 mice, **p<0.01, unpaired t test). Supplementary Figure 2: LPS treated Podocin-Cre Klf15flox/flox mice exhibit a lack of recovery in proteinaceous casts and tubular dilatation after DEX administration. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

AP-4 Mediates Export of ATG9A from the Trans-Golgi Network to Promote
AP-4 mediates export of ATG9A from the trans-Golgi PNAS PLUS network to promote autophagosome formation Rafael Matteraa,1, Sang Yoon Parka,1, Raffaella De Pacea, Carlos M. Guardiaa, and Juan S. Bonifacinoa,2 aCell Biology and Neurobiology Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892 Edited by Pietro De Camilli, Howard Hughes Medical Institute and Yale University, New Haven, CT, and approved November 6, 2017 (received for review October 2, 2017) AP-4 is a member of the heterotetrameric adaptor protein (AP) mediated by a noncanonical YRYRF sequence in the receptor- complex family involved in protein sorting in the endomembrane associated, transmembrane AMPA receptor regulatory proteins system of eukaryotic cells. Interest in AP-4 has recently risen with (TARPs) (17). Finally, in the δ2 glutamate receptor protein, the discovery that mutations in any of its four subunits cause a binding to μ4 depends on several phenylalanine residues in a form of hereditary spastic paraplegia (HSP) with intellectual noncanonical context (18). disability. The critical sorting events mediated by AP-4 and the Interest in AP-4 has recently risen because of the discovery of pathogenesis of AP-4 deficiency, however, remain poorly under- mutations in genes encoding each of the subunits of AP-4 in stood. Here we report the identification of ATG9A, the only a subset of autosomal recessive hereditary spastic paraplegias multispanning membrane component of the core autophagy ma- (HSPs), namely, SPG47 (AP4B1/β4), SPG50 (AP4M1/μ4), SPG51 chinery, as a specific AP-4 cargo. AP-4 promotes signal-mediated (AP4E1/e), and SPG52 (AP4S1/σ4) (19–21). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Myopathy Genes (HGNC) Neuropathy (HGNC) Neuromuscular Disease
Myopathy Genes Neuropathy Neuromuscular Disease (HGNC) (HGNC) (HGNC) ABHD5 ABCA1 ADCK3 ACTG2 ACO2 AGRN AGK AGXT ALS2 ALDOA AIFM1 ANG AMER1 ALAD AP4B1 ANO5 AMACR AP4E1 AR AP1S1 AP4M1 AUH APTX AP4S1 B4GALT1 AR AP5Z1 CACNA1S ATL3 ATM CASQ1 B4GALNT1 ATXN10 CCDC78 BAG3 ATXN7 CHCHD10 BRP44L BEAN1 CHRNA1 C12orf65 C9orf72 CHRNB1 C19orf12 CACNB4 CHRND C1NH CAPN3 CHRNE CECR1 CHAT CLPB CISD2 CHKB COL6A1 CLCF1 CHMP2B COL6A2 CLCN2 CHRNG COL6A3 CLP1 CLCN1 COLQ CMT2G COL9A3 CTNS CMT2H COQ2 DGUOK CMTDIA COQ6 DNA2 CMTX2 COQ9 DNAJB6 CMTX3 COX15 DNAJC19 COASY CPT1A DNM2 COX6A1 CYP7B1 DPM2 CPOX DAG1 DYSF CYP27A1 DDHD2 EMD CYP2U1 DOK7 EPG5 DARS2 DPAGT1 FAM111B DCAF8 DPM3 FBXL4 DDHD1 DUX4 FKBP14 DFNX5 ECEL1 FKRP DHTKD1 ERBB3 FLH1 DIAPH3 ERLIN2 FLNC DNAJB2 FA2H HNRNPA1 DNAJC3 FKTN HNRNPDL ELOVL5 FUS HNRPA2B1 ERCC8 G6PC KLHL40 FAH GFPT1 KLHL41 FAM126A GLE1 LAMA2 FBN1 GYS2 LDB3 FMR1 HSPD1 LMOD3 FXN IFRD1 MEGF10 GALC INF2 MGME1 GBE1 ISPD MTAP GJC2 ITGA7 MTMR14 GP1BA ITPR1 MYF6 HADHA KCNA1 MYH14 HADHB KCNC3 MYLK2 HFE KCNE3 NARS2 HINT1 KCNJ18 NEB HK1 KCNJ2 ORAI1 HMBS KIAA0196 PRKAG2 HSD17B4 KIF21A PTEN HSN1B L1CAM RBCK1 IARS2 LAMB2 RET IGHMBP2 LARGE RMND1 KCNJ10 MCCC2 SCN4A KIF5A MRE11A SERAC1 LRSAM1 MRPL3 SGCA LYST MTO1 SIL1 MANBA MTPAP SPEG MARS MTTP STAC3 MTATP6 MUSK STIM1 MYH14 MYBPC3 SYNE1 MYOT MYH3 SYNE2 NAMSD MYH8 TAZ NF2 NF1 TIA1 NGLY1 NIPA1 TMEM43 NMSR NOP56 TNPO3 NOTCH3 OPTN TNXB OPA1 PDSS2 TPM2 OPA3 PDYN TRPV4 OTOF PFN1 UBA1 PDK3 PHKA2 VCP PDSS1 PHKG2 XDH PEX10 PHOX2A ACADS PEX2 PIP5K1C ACADVL PMM2 PLEC ACTA1 PNPLA6 PLP1 AGL PPOX POMGNT1 AMPD1 PRICKLE1 -

GENETIC TESTING REQUISITION Please Ship All
GENETIC TESTING REQUISITION 1-844-363-4357· [email protected] Schillingallee 68 · 18057 Rostock Germany Attention Patient: Please visit your nearest LifeLabs or CML Healthcare Patient Service Centre for sample collection LL: K012-01/ CML: CEN CONTRACT # Report to Physician Billing # LifeLabs Demographic Ordering Physician Name Label Physician Signature: Ordering Physician Address: Tel: Fax: Address & Contact Info: Copy to (name & contact info): Name: Contact: Bill to Contract # K012-01 (patient does not pay at time of collection) Patient Gender: (M/F) Patient Name (Last, First): Patient DOB: (YYYY/MM/DD) Patient Address: Patient Health Card: Patient Telephone: Please ship all NON-PRENATAL samples to: LifeLabs · Attn CDS Department • 100 International Boulevard• Toronto ON• M9W6J6 TEST REQUESTED LL TR # / CML TC# □ Genetic Test - Blood Sample 2 x 4mL EDTA 4005 □ Genetic Test (Pediatric) - Blood Sample 1 x 2mL EDTA 4008 □ Genetic Test - Other Sample Type 4014 PRENATAL SAMPLES: Please ship directly to CENTOGENE. Date Blood Collected (YYYY/MM/DD): ___________ Time Blood Collected (HH:MM)) :________ Collector Name: ___________________ GENETIC TESTING CONSENT I understand that a DNA specimen will be sent to LifeLabs for genetic testing. My physician has told me about the condition(s) being tested and its genetic basis. I am aware that correct information about the relationships between my family members is important. I agree that my specimen and personal health information may be sent to Centogene AG at their lab in Germany (address below). To ensure accurate testing, I agree that the results of any genetic testing that I have had previously completed by Centogene AG may be shared with LifeLabs. -

Mutation in the AP4M1 Gene Provides a Model for Neuroaxonal Injury in Cerebral Palsy
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector ARTICLE Mutation in the AP4M1 Gene Provides a Model for Neuroaxonal Injury in Cerebral Palsy Annemieke J.M.H. Verkerk,1 Rachel Schot,2 Belinda Dumee,1 Karlijn Schellekens,1 Sigrid Swagemakers,1 Aida M. Bertoli-Avella,2 Maarten H. Lequin,3 Jeroen Dudink,4 Paul Govaert,4 A.L. van Zwol,4 Jennifer Hirst,5 Marja W. Wessels,2 Coriene Catsman-Berrevoets,6 Frans W. Verheijen,2 Esther de Graaff,2 Irenaeus F.M. de Coo,6 Johan M. Kros,7 Rob Willemsen,2 Patrick J. Willems,8 Peter J. van der Spek,1 and Grazia M.S. Mancini2,* Cerebral palsy due to perinatal injury to cerebral white matter is usually not caused by genetic mutations, but by ischemia and/or inflam- mation. Here, we describe an autosomal-recessive type of tetraplegic cerebral palsy with mental retardation, reduction of cerebral white matter, and atrophy of the cerebellum in an inbred sibship. The phenotype was recorded and evolution followed for over 20 years. Brain lesions were studied by diffusion tensor MR tractography. Homozygosity mapping with SNPs was performed for identification of the chromosomal locus for the disease. In the 14 Mb candidate region on chromosome 7q22, RNA expression profiling was used for selecting among the 203 genes in the area. In postmortem brain tissue available from one patient, histology and immunohistochemistry were performed. Disease course and imaging were mostly remi- niscent of hypoxic-ischemic tetraplegic cerebral palsy, with neuroaxonal degeneration and white matter loss. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Full Disclosure Form Information Provided by the Authors Is Available with the Full Text of This Article at Neurology.Org/NG
ARTICLE OPEN ACCESS AP4 deficiency A novel form of neurodegeneration with brain iron accumulation? Agathe Roubertie, MD, PhD, Nelson Hieu, MSc,* Charles-Joris Roux, MD,* Nicolas Leboucq, MD, Correspondence Gael Manes, PhD, Majida Charif, PhD, Bernard Echenne, MD, Cyril Goizet, MD, PhD, Claire Guissart, PhD, Dr. Roubertie [email protected] Pierre Meyer, MD, Cecilia Marelli, MD, François Rivier, MD, PhD, Lydie Burglen, MD, Rita Horvath, MD, PhD, Christian P. Hamel, MD, PhD, and Guy Lenaers, PhD Neurol Genet 2018;4:e217. doi:10.1212/NXG.0000000000000217 Abstract Objective To describe the clinico-radiological phenotype of 3 patients harboring a homozygous novel AP4M1 pathogenic mutation. Methods The 3 patients from an inbred family who exhibited early-onset developmental delay, tetra- paresis, juvenile motor function deterioration, and intellectual deficiency were investigated by magnetic brain imaging using T1-weighted, T2-weighted, T2*-weighted, fluid-attenuated in- version recovery, susceptibility weighted imaging (SWI) sequences. Whole-exome sequencing was performed on the 3 patients. Results In the 3 patients, brain imaging identified the same pattern of bilateral SWI hyposignal of the globus pallidus, concordant with iron accumulation. A novel homozygous nonsense mutation was identified in AP4M1, segregating with the disease and leading to truncation of half of the adap domain of the protein. Conclusions Our results suggest that AP4M1 represents a new candidate gene that should be considered in the neurodegeneration with brain -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7, -
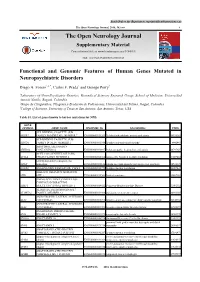
The Open Neurology Journal, 2016, 10, I-Xv I the Open Neurology Journal Supplementary Material Content List Available At
Send Orders for Reprints to [email protected] The Open Neurology Journal, 2016, 10, i-xv i The Open Neurology Journal Supplementary Material Content list available at: www.benthamopen.com/TONEUJ/ DOI: 10.2174/1874205X01610010143 Functional and Genomic Features of Human Genes Mutated in Neuropsychiatric Disorders Diego A. Forero1,4, *, Carlos F. Prada2 and George Perry3 1Laboratory of NeuroPsychiatric Genetics, Biomedical Sciences Research Group, School of Medicine, Universidad Antonio Nariño. Bogotá, Colombia 2Grupo de Citogenética, Filogenia y Evolución de Poblaciones, Universidad del Tolima. Ibagué, Colombia 3College of Sciences, University of Texas at San Antonio, San Antonio, Texas, USA Table S1. List of genes known to harbor mutations for NPD. GENE SYMBOL GENE NAME ENSEMBL ID DISORDERS PMID ATP-BINDING CASSETTE, SUB- ABCB7 FAMILY B (MDR/TAP), MEMBER 7 ENSG00000131269 X-linked sideroblastic anemia and ataxia 10196363 ATP-BINDING CASSETTE, SUB- ABCD1 FAMILY D (ALD), MEMBER 1 ENSG00000101986 X-linked adrenoleukodystrophy 8441467 ABHYDROLASE DOMAIN ABHD12 CONTAINING 12 ENSG00000100997 Polyneuropathy, hearing loss and ataxia 20797687 ACYL-COA SYNTHETASE LONG- ACSL4 CHAIN FAMILY MEMBER 4 ENSG00000068366 nonspecific X-linked mental retardation 11889465 activity-dependent neuroprotector ADNP homeobox ENSG00000101126 autism spectrum disorder and intellectual disability 24531329 AGTR2 ANGIOTENSIN II RECEPTOR, TYPE 2 ENSG00000180772 X-linked mental retardation 12089445 ABELSON HELPER INTEGRATION AHI1 SITE 1 ENSG00000135541 Joubert -

A Novel AP4M1 Mutation in Autosomal Recessive Cerebral Palsy Syndrome
Jameel et al. BMC Medical Genetics 2014, 15:133 http://www.biomedcentral.com/1471-2350/15/133 RESEARCH ARTICLE Open Access A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency Muhammad Jameel1, Joakim Klar2, Muhammad Tariq1, Abubakar Moawia1, Naveed Altaf Malik1, Syeda Seema Waseem1, Uzma Abdullah1, Tahir Naeem Khan1, Raili Raininko3, Shahid Mahmood Baig1 and Niklas Dahl2* Abstract Background: Cerebral palsy (CP) is a heterogeneous neurodevelopmental disorder associated with intellectual disability in one-third of cases. Recent findings support Mendelian inheritance in subgroups of patients with the disease. The purpose of this study was to identify a novel genetic cause of paraplegic CP with intellectual disability in a consanguineous Pakistani family. Methods: We performed whole-exome sequencing (WES) in two brothers with CP and intellectual disability. Analysis of AP4M1 mRNA was performed using quantitative real-time PCR on total RNA from cultured fibroblasts. The brothers were investigated clinically and by MRI. Results: We identified a novel homozygous AP4M1 mutation c.194_195delAT, p.Y65Ffs*50 in the affected brothers. Quantitative RT-PCR analysis showed markedly reduced AP4M1 mRNA levels suggesting partial non-sense mediated mRNA decay. Several clinical and MRI features were consistent with AP-4 complex deficiency. However, in contrast to previously reported cases with AP4M1 mutations our patients show an aggressive behavior and a relatively late onset of disease. Conclusion: This study shows an AP4M1 mutation associated with aggressive behavior in addition to mild dysmorphic features, intellectual disability, spastic paraparesis and reduced head circumference. Our findings expand the clinical spectrum associated with AP-4 complex deficiency and the study illustrates the importance of MRI and WES in the diagnosis of patients with CP and intellectual disability.