Network Analysis Identifies Sex-Specific Gene Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ZFX Acts As a Transcriptional Activator in Multiple Types of Human Tumors by Binding Downstream from Transcription Start Sites at the Majority of Cpg Island Promoters
Downloaded from genome.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press Research ZFX acts as a transcriptional activator in multiple types of human tumors by binding downstream from transcription start sites at the majority of CpG island promoters Suhn Kyong Rhie, Lijun Yao, Zhifei Luo, Heather Witt, Shannon Schreiner, Yu Guo, Andrew A. Perez, and Peggy J. Farnham Department of Biochemistry and Molecular Medicine and the Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, Los Angeles, California 90089, USA High expression of the transcription factor ZFX is correlated with proliferation, tumorigenesis, and patient survival in mul- tiple types of human cancers. However, the mechanism by which ZFX influences transcriptional regulation has not been determined. We performed ChIP-seq in four cancer cell lines (representing kidney, colon, prostate, and breast cancers) to identify ZFX binding sites throughout the human genome. We identified roughly 9000 ZFX binding sites and found that most of the sites are in CpG island promoters. Moreover, genes with promoters bound by ZFX are expressed at higher levels than genes with promoters not bound by ZFX. To determine if ZFX contributes to regulation of the promoters to which it is bound, we performed RNA-seq analysis after knockdown of ZFX by siRNA in prostate and breast cancer cells. Many genes with promoters bound by ZFX were down-regulated upon ZFX knockdown, supporting the hypothesis that ZFX acts as a transcriptional activator. Surprisingly, ZFX binds at +240 bp downstream from the TSS of the responsive pro- moters. Using Nucleosome Occupancy and Methylome Sequencing (NOMe-seq), we show that ZFX binds between the open chromatin region at the TSS and the first downstream nucleosome, suggesting that ZFX may play a critical role in pro- moter architecture. -

Differential Expression of ZFX Gene in Gastric Cancer
Differential expression of ZFX gene in gastric cancer 1 2,3, 1 PARVANEH NIKPOUR , MODJTABA EMADI-BAYGI *, FAEZEH MOHAMMAD-HASHEM , MOHAMAD REZA MARACY4 and SHAGHAYEGH HAGHJOOY-JAVANMARD5 1Department of Genetics and Molecular Biology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran 2Department of Genetics, Faculty of Basic Sciences, 3Research Institute of Biotechnology, Shahrekord University, Shahrekord, Iran 4Department of Community Medicine, Faculty of Medicine, 5Applied Physiology Research Center, Isfahan University of Medical Sciences, Isfahan, Iran *Corresponding author (Fax, +98-311-7753480; Email, [email protected]) Gastric cancer accounts for 8% of the total cancer cases and 10% of total cancer deaths worldwide. In Iran, gastric cancer is the leading cause of national cancer-related mortality. Most human cancers show substantial heterogeneity. The cancer stem cell (CSC) hypothesis has been proposed to reconcile this heterogeneity. ZFX encodes a member of the krueppel C2H2-type zinc-finger protein family that is required as a transcriptional regulator for self-renewal of stem cells. A total of 30 paired tissue gastric samples were examined for ZFX gene expression by quantitative real-time RT-PCR. Although the relative expression of the gene was significantly high in 47% of the examined tumour tissues, its expression was low in the others (53%). There was a statistically significant association between the ZFX gene expression and different tumour types and grades. This is the first report that shows ZFX was differentially expressed in gastric cancer. Of note, it was overexpressed in diffused-type and grade III gastric tumoural tissues. Due to this, ZFX may have the potential to be used as a target for therapeutic interventions. -

Genetics of Azoospermia
International Journal of Molecular Sciences Review Genetics of Azoospermia Francesca Cioppi , Viktoria Rosta and Csilla Krausz * Department of Biochemical, Experimental and Clinical Sciences “Mario Serio”, University of Florence, 50139 Florence, Italy; francesca.cioppi@unifi.it (F.C.); viktoria.rosta@unifi.it (V.R.) * Correspondence: csilla.krausz@unifi.it Abstract: Azoospermia affects 1% of men, and it can be due to: (i) hypothalamic-pituitary dysfunction, (ii) primary quantitative spermatogenic disturbances, (iii) urogenital duct obstruction. Known genetic factors contribute to all these categories, and genetic testing is part of the routine diagnostic workup of azoospermic men. The diagnostic yield of genetic tests in azoospermia is different in the different etiological categories, with the highest in Congenital Bilateral Absence of Vas Deferens (90%) and the lowest in Non-Obstructive Azoospermia (NOA) due to primary testicular failure (~30%). Whole- Exome Sequencing allowed the discovery of an increasing number of monogenic defects of NOA with a current list of 38 candidate genes. These genes are of potential clinical relevance for future gene panel-based screening. We classified these genes according to the associated-testicular histology underlying the NOA phenotype. The validation and the discovery of novel NOA genes will radically improve patient management. Interestingly, approximately 37% of candidate genes are shared in human male and female gonadal failure, implying that genetic counselling should be extended also to female family members of NOA patients. Keywords: azoospermia; infertility; genetics; exome; NGS; NOA; Klinefelter syndrome; Y chromosome microdeletions; CBAVD; congenital hypogonadotropic hypogonadism Citation: Cioppi, F.; Rosta, V.; Krausz, C. Genetics of Azoospermia. 1. Introduction Int. J. Mol. Sci. -

ZFX Rabbit Polyclonal Antibody – TA343385 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA343385 ZFX Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: WB Reactivity: Human Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: The immunogen for Anti-ZFX antibody is: synthetic peptide directed towards the C-terminal region of Human ZFX. Synthetic peptide located within the following region: TTDASGFKRHVISIHTKDYPHRCEYCKKGFRRPSEKNQHIMRHHKEVGLP Formulation: Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose. Note that this product is shipped as lyophilized powder to China customers. Purification: Affinity Purified Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 83 kDa Gene Name: zinc finger protein, X-linked Database Link: NP_003401 Entrez Gene 7543 Human P17010 Background: The function of this protein remains unknown. Synonyms: ZNF926 Note: Immunogen Sequence Homology: Dog: 100%; Pig: 100%; Rat: 100%; Horse: 100%; Human: 100%; Mouse: 100%; Bovine: 100%; Rabbit: 100%; Zebrafish: 100%; Guinea pig: 100% Protein Families: Transcription Factors This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 ZFX Rabbit Polyclonal Antibody – TA343385 Product images: Host: Rabbit; Target Name: ZFX; Sample Tissue: ACHN Whole cell lysates; Antibody Dilution: 1.0 ug/ml This product is to be used for laboratory only. -
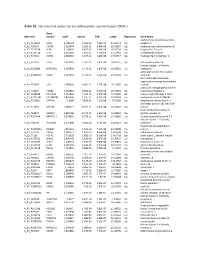
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Cpg Island-Mediated Global Gene Regulatory Modes in Mouse Embryonic Stem Cells
ARTICLE Received 26 Mar 2014 | Accepted 3 Oct 2014 | Published 18 Nov 2014 DOI: 10.1038/ncomms6490 OPEN CpG island-mediated global gene regulatory modes in mouse embryonic stem cells Samuel Beck1, Bum-Kyu Lee1, Catherine Rhee1, Jawon Song2, Andrew J. Woo3 & Jonghwan Kim1,4,5 Both transcriptional and epigenetic regulations are fundamental for the control of eukaryotic gene expression. Here we perform a compendium analysis of 4200 large sequencing data sets to elucidate the regulatory logic of global gene expression programs in mouse embryonic stem (ES) cells. We define four major classes of DNA-binding proteins (Core, PRC, MYC and CTCF) based on their target co-occupancy, and discover reciprocal regulation between the MYC and PRC classes for the activity of nearly all genes under the control of the CpG island (CGI)-containing promoters. This CGI-dependent regulatory mode explains the functional segregation between CGI-containing and CGI-less genes during early development. By defining active enhancers based on the co-occupancy of the Core class, we further demon- strate their additive roles in CGI-containing gene expression and cell type-specific roles in CGI-less gene expression. Altogether, our analyses provide novel insights into previously unknown CGI-dependent global gene regulatory modes. 1 Department of Molecular Biosciences, The University of Texas at Austin, Austin, Texas 78712, USA. 2 Texas Advanced Computing Center, The University of Texas at Austin, Austin, Texas 78758, USA. 3 School of Medicine and Pharmacology, Royal Perth Hospital Unit, The University of Western Australia, Perth, WA 6000, Australia. 4 Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, Texas 78712, USA. -

Expression of ZFX Gene Correlated with the Central Features of the Neoplastic Phenotype in Human Brain Tumors with Distinct Phenotypes
Original Article Expression of ZFX gene correlated with the central features of the neoplastic phenotype in human brain tumors with distinct phenotypes Azita Afzali, Modjtaba Emadi‑Baygi, Parvaneh Nikpour1, Fatemehe Nazemroaya1, Majid Kheirollahi1 Department of Genetics, Faculty of Basic Sciences, Shahrekord University, Shahrekord, 1Department of Genetics and Molecular Biology, School of Medicine, Pediatric Inherited Diseases Research Center, Research Institute for Primordial Prevention of Non-communicable Disease, Isfahan University of Medical Sciences, Isfahan, Iran Abstract Background: The zinc finger transcription factor zinc finger protein, X‑linked (ZFX) acts as an important director of self‑renewal in several stem cell types. Moreover, ZFX expression abnormally increases in various cancers and relates to tumor grade. We performed this study, to examine its role in the pathogenesis of astrocytoma and meningioma. Materials and Methods: We used real‑time reverse transcription polymerase chain reaction method for evaluation of ZFX expression in 25 astrocytoma tumoral tissue and 25 meningioma tumoral tissues with different WHO grades. Furthermore, the association of gene expression with various clinic‑pathological characteristics was examined. Results: We found that there is a significant association between gene expression and different tumor grades, the presence or absence of invasion, forming and nonforming of glomeruloid vessels, the age over or under 50 and the presence or absence of calcification in astrocytomas. This is the first report that shows that ZFX was directly correlated with the central features of the neoplastic phenotype, including the growth of cancer cells, angiogenesis, and invasion. Conclusion: Regarding all the above‑mentioned studies, it is highly plausible that silencing the expression of ZFX gene in gliomas has a major role in the therapeutic interventions of the disease in future. -

Evolutionary Strata on the Mouse X Chromosome Correspond to Strata on the Human X Chromosome Sara A
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Evolutionary Strata on the Mouse X Chromosome Correspond to Strata on the Human X Chromosome Sara A. Sandstedt1 and Priscilla K. Tucker Department of Ecology and Evolutionary Biology, and Museum of Zoology, University of Michigan, Ann Arbor, Michigan 48109, USA Lahn and Page previously observed that genes on the human X chromosome were physically arranged along the chromosome in “strata,” roughly ordered by degree ofdivergence fromrela ted genes on the Y chromosome. They hypothesized that this ordering results from a historical series of suppressions ofrecombination along the mammalian Y chromosome, thereby allowing formerly recombining X and Y chromosomal genes to diverge independently. Here predictions ofthis hypothesis are confirmedin a non primate mammalian order, Rodentia, through an analysis ofeight gene pairs fromthe X and Y chromosomes ofthe ho use mouse, Mus musculus. The mouse X chromosome has been rearranged relative to the human X, so strata were not found in the same physical order on the mouse X. However, based on synonymous evolutionary distances, X-linked genes in M. musculus fall into the same strata as orthologous genes in humans, as predicted. The boundary between strata 2 and 3 is statistically significant, but the boundary between strata 1 and 2 is not significant in mice. An analysis ofsmaller fragmentsofSmcy, Smcx, Zfy, and Zfx from seven species of Mus confirmed that the strata in Mus musculus were representative ofthe genus Mus. [Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. -

Probing the Gene Expression Database for Candidate Genes
European Journal of Human Genetics (1999) 7, 910–919 t © 1999 Stockton Press All rights reserved 1018–4813/99 $15.00 http://www.stockton-press.co.uk/ejhg ARTICLE Probing the Gene eXpression Database for candidate genes Maurice AM van Steensel, J Celli, JH van Bokhoven and HG Brunner Department of Human Genetics, University Hospital Nijmegen, The Netherlands We report on a strategy for the identification of candidate genes for multiple malformation syndromes using expression data available in public databases. The basis for this pilot study was the assumption that, for a multiple malformation syndrome, the expression pattern of the causative gene should at least cover the organs or tissues affected by the syndrome. Twenty malformation syndromes were selected from the OMIM and defined by three to five main symptoms. These key symptoms were translated into anatomical terms that were used to query the Gene eXpression Database (GXD). The searches covered 65% of the database and yielded an average of 16 candidate genes per syndrome. Of these, 23% were ubiquitously expressed or housekeeping genes. Further database evaluation of these potential candidate genes was based on positional information and on information from mouse knockouts. In a first experiment, the correct gene was identified as a candidate in four of seven syndromes for which the causative gene is already known. In addition, this strategy identified new candidate genes for disorders for which the genetic basis is unknown. We identified candidate genes for the Walker-Warburg, DOOR, C, scalp–ear–nipple and oculocerebral hypopigmentation syndromes. Our results suggest that it may ultimately be feasible to identify disease genes by probing gene expression databases with simple syndrome descriptions. -

Comparison of Human ZFY and ZFX Transcripts (Sex Determination/"Zinc Rmgers"/X Chromosome/Y Chromosome/Polymerase Chain Reaction) MARK S
Proc. Nati. Acad. Sci. USA Vol. 87, pp. 1681-1685, March 1990 Genetics Comparison of human ZFY and ZFX transcripts (sex determination/"zinc rmgers"/X chromosome/Y chromosome/polymerase chain reaction) MARK S. PALMER, PHILIPPE BERTA*, ANDREW H. SINCLAIR, BARBARA PYM, AND PETER N. GOODFELLOW Human Molecular Genetics, Imperial Cancer Research Fund, Lincoln's Inn Fields, London WC2A 3PX, United Kingdom Communicated by M. F. Lyon, November 16, 1989 ABSTRACT ZFY is a candidate for the primary sex- Southern Blotting. Endonuclease-digested DNA was elec- determining gene (TDF, testis-determining factor) on the hu- trophoresed in 0.8% agarose gels and transferred and fixed to man Y chromosome. We have isolated cDNA clones ofZFY and Hybond-N+ filters (Amersham) (10). its homologue on the X chromosome, ZFX. The transcripts of Hybridization. (i) Oligonucleotide probes. Oligonucleotide these genes are very similar to each other and encode predicted 1818 was labeled with 32P at its 5' end with phage T4 proteins of equal size. The conceptual amino acid sequence of polynucleotide kinase. Library filters were hybridized in 5 x both proteins contains an acidic domain, similar to the acti- SSC/20 mM sodium phosphate, pH 7.0/lOx Denhardt's vation domain of transcription factors, and a potential nucleic solution/10%o dextran sulfate/7% SDS containing oligonu- acid-binding domain of 13 "zinc ringers."' We have used the cleotide (106 cpm/ml) and heterologous DNA (100,ug/ml). polymerase chain reaction to demonstrate the expression of (ii) Plasmidprobes. Plasmid inserts were radiolabeled by the ZFY and ZFX in a wide range of adult and fetal human tissues random primer method (11). -

Sex-Chromosome Dosage Effects on Gene Expression in Humans
Sex-chromosome dosage effects on gene expression in humans Armin Raznahana,1, Neelroop N. Parikshakb,c, Vijay Chandrand, Jonathan D. Blumenthala, Liv S. Clasena, Aaron F. Alexander-Blocha, Andrew R. Zinne,f, Danny Wangsag, Jasen Wiseh, Declan G. M. Murphyi, Patrick F. Boltoni, Thomas Riedg, Judith Rossj, Jay N. Gieddk, and Daniel H. Geschwindb,c aDevelopmental Neurogenomics Unit, National Institute of Mental Health, National Institutes of Health, Bethesda, MD 20892; bNeurogenetics Program, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095; cCenter for Autism Research and Treatment, Semel Institute, David Geffen School of Medicine, University of California, Los Angeles, CA 90095; dDepartment of Pediatrics, School of Medicine, University of Florida, Gainesville, FL 32610; eMcDermott Center for Human Growth and Development, University of Texas Southwestern Medical School, Dallas, TX 75390; fDepartment of Internal Medicine, University of Texas Southwestern Medical School, Dallas, TX 75390; gGenetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892; hQiagen, Frederick, MD 21703; iInstitute of Psychiatry, Psychology and Neuroscience, King’s College London, University of London, London WC1B 5DN, United Kingdom; jDepartment of Pediatrics, Thomas Jefferson University, Philadelphia, PA 19107; and kDepartment of Psychiatry, University of California, San Diego, La Jolla, CA 92093 Edited by James A. Birchler, University of Missouri, -

Cell Culture, Sex Determination and Single Cell Cloning of Ovine Transgenic Satellite Cells in Vitro Fatemeh Salabi1, Mahmood Nazari2 and Wen G Cao1*
Salabi et al. Journal of Biological Research-Thessaloniki (2014) 21:22 DOI 10.1186/s40709-014-0022-z RESEARCH Open Access Cell culture, sex determination and single cell cloning of ovine transgenic satellite cells in vitro Fatemeh Salabi1, Mahmood Nazari2 and Wen G Cao1* Abstract Background: This study was performed to describe the basic methods to isolate and culture of primary satellite cells (PSCs) obtained from 50 to 60-day-old sheep fetuses, single cell cloning of transfected PSCs and sexing of ovine PSCs based on the ZFY/ZFX, amelogenin and high-motility-group (HMG) box sequences. Results: Three-step enzymatic digestion method increased PSCs isolation from tissue and reduced the damage of cells during long time incubation with enzymes. The results of cloning showed that the 103 and 81 clones (from a total of 184 clones) were derived from feeder and bFGF treatment, respectively. The overall sexing efficiency in the present study was 100%. Southern blot results of sex determination were in complete agreement with PCR-amplified bands which confirmed that the HMG box of SRY gene amplified from the ovine genome and that was specific for male. Conclusions: We successfully isolated and cultured sheep primary satellite cells via mechanical and enzymatic disaggregation. Our finding demonstrated that use of feeder and addition of bFGF to the culture medium improved cloning efficiency. The results of sex detection demonstrated that these methods can be applied to detect the sex of primary satellite cells and to determine the sex of sheep embryo prior to produce sheep embryos by somatic cell nuclear transfer technique in vitro.