Supplementary Materials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

C8cc08685k1.Pdf
Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2018 Supporting Information Minimalist Linkers Suitable for Irreversible Inhibitors in Simultaneous Proteome Profiling, Live-Cell Imaging and Drug Screening Cuiping Guo,Yu Chang, Xin Wang, Chengqian Zhang, Piliang Hao*, Ke Ding and Zhengqiu Li* School of Pharmacy, Jinan University, Guangzhou, China 510632 *Corresponding author ([email protected]) 1. General Information All chemicals were purchased from commercial vendors and used without further purification, unless indicated otherwise. All reactions requiring anhydrous conditions were carried out under argon or nitrogen atmosphere using oven-dried glassware. AR-grade solvents were used for all reactions. Reaction progress was monitored by TLC on pre-coated silica plates (Merck 60 F254 nm, 0.25 µm) and spots were visualized by UV, iodine or other suitable stains. Flash column chromatography was carried out using silica gel (Qingdao Ocean). All NMR spectra (1H-NMR, 13C-NMR) were recorded on Bruker 300 MHz/400 MHz NMR spectrometers. Chemical shifts were reported in parts per million (ppm) referenced with respect to appropriate internal standards or residual solvent peaks (CDCl3 = 7.26 ppm, DMSO-d6 = 2.50 ppm). The following abbreviations were used in reporting spectra, br s (broad singlet), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets). Mass spectra were obtained on Agilent LC-ESI-MS system. All analytical HPLC were carried out on Agilent system. Water with 0.1% TFA and acetonitrile with 0.1% TFA were used as eluents and the flow rate was 0.5 mL/min. -

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

Proteínas De Superfície De Paracoccidioides Brasiliensis
UNIVERSIDADE DE BRASÍLIA FACULDADE DE MEDICINA PROGRAMA DE PÓS-GRADUAÇÃO EM PATOLOGIA MOLECULAR Proteínas de superfície de Paracoccidioides brasiliensis CANDIDATA: NADYA DA SILVA CASTRO ORIENTADORA: DRA. CÉLIA MARIA DE ALMEIDA SOARES TESE APRESENTADA AO PROGRAMA DE PÓS-GRADUAÇÃO EM PATOLOGIA MOLECULAR, DA FACULDADE DE MEDICINA, DA UNIVERSIDADE DE BRASÍLIA COMO REQUISITO PARCIAL À OBTENÇÃO DO TÍTULO DE DOUTOR EM PATOLOGIA MOLECULAR. BRASÍLIA – DF MAIO 2008 TRABALHO REALIZADO NO LABORATÓRIO DE BIOLOGIA MOLECULAR, DEPARTAMENTO DE BIOQUÍMICA E BIOLOGIA MOLECULAR, INSTITUTO DE CIÊNCIAS BIOLÓGICAS, DA UNIVERSIDADE FEDERAL DE GOIÁS. APOIO FINANCEIRO: CAPES/ CNPQ/ FINEP/ FAPEG/ SECTEC-GO. II BANCA EXAMINADORA TITULARES Profa. Dra. Célia Maria de Almeida Soares, Instituto de Ciências Biológicas, Universidade Federal de Goiás. Prof. Dr. Augusto Schrank Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul Prof. Dr. Ivan Torres Nicolau de Campos Instituto de Ciências Biológicas, Universidade Federal de Goiás. Prof. Dr. Bergmann Morais Ribeiro Instituto de Ciências Biológicas, Universidade de Brasília. Prof. Dra. Anamélia Lorenzetti Bocca Instituto de Ciências Biológicas, Universidade de Brasília. SUPLENTE Prof. Dr. Fernando Araripe Gonçalves Torres Instituto de Ciências Biológicas, Universidade de Brasília. III ´- Os homens do seu planeta ² disse o pequeno Príncipe ² cultivam cinco mil rosas num jardim... e não encontram o que procuram... - É verdade ² respondi. - E, no entanto, o que eles procuram poderia ser encontrado numa só rosa, ou num pouco de água... - É verdade. E o principezinho acrescentou: Mas os olhos são cegos. eSUHFLVRYHUFRPRFRUDomRµ ³O pequeno príncipe´ de Antonie de Saint-Exupéry IV Dedico esta tese aos meus queridos pais, Nadson e Genialda, que foram e são exemplos de dedicação e de perseverança e cujos incentivos, apoio e amor contribuíram em muito para a realização deste trabalho. -

Structure of the Mammalian 80S Initiation Complex with Initiation Factor 5B on HCV-IRES RNA
ARTICLES Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA Hiroshi Yamamoto1,3, Anett Unbehaun1,3, Justus Loerke1, Elmar Behrmann1, Marianne Collier1, Jörg Bürger1,2, Thorsten Mielke1,2 & Christian M T Spahn1 The universally conserved eukaryotic initiation factor (eIF) 5B, a translational GTPase, is essential for canonical translation initiation. It is also required for initiation facilitated by the internal ribosomal entry site (IRES) of hepatitis C virus (HCV) RNA. Met eIF5B promotes joining of 60S ribosomal subunits to 40S ribosomal subunits bound by initiator tRNA (Met-tRNAi ). However, the exact molecular mechanism by which eIF5B acts has not been established. Here we present cryo-EM reconstructions of the Met mammalian 80S–HCV-IRES–Met-tRNAi –eIF5B–GMPPNP complex. We obtained two substates distinguished by the rotational state of the ribosomal subunits and the configuration of initiator tRNA in the peptidyl (P) site. Accordingly, a combination of Met conformational changes in the 80S ribosome and in initiator tRNA facilitates binding of the Met-tRNAi to the 60S P site and redefines the role of eIF5B as a tRNA-reorientation factor. Met Eukaryotic translation initiation is a highly regulated process within stable ribosomal binding of Met-tRNAi and elongation competence the translation cycle that proceeds via 48S and 80S initiation-complex in vivo11–13. However, the molecular mechanism linking eIF5B–GTP Met Met intermediates. At least 12 eIFs facilitate recruitment of Met-tRNAi hydrolysis and proper placement of Met-tRNAi in the ribosomal and mRNA to the ribosomal 40S subunit and regulate the interaction of P site is not known. -

Reprogramming of Trna Modifications Controls the Oxidative Stress Response by Codon-Biased Translation of Proteins
Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Chan, Clement T.Y. et al. “Reprogramming of tRNA Modifications Controls the Oxidative Stress Response by Codon-biased Translation of Proteins.” Nature Communications 3 (2012): 937. As Published http://dx.doi.org/10.1038/ncomms1938 Publisher Nature Publishing Group Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/76775 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins Clement T.Y. Chan,1,2 Yan Ling Joy Pang,1 Wenjun Deng,1 I. Ramesh Babu,1 Madhu Dyavaiah,3 Thomas J. Begley3 and Peter C. Dedon1,4* 1Department of Biological Engineering, 2Department of Chemistry and 4Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; 3College of Nanoscale Science and Engineering, University at Albany, SUNY, Albany, NY 12203 * Corresponding author: PCD, Department of Biological Engineering, NE47-277, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139; tel 617-253-8017; fax 617-324-7554; email [email protected] 2 ABSTRACT Selective translation of survival proteins is an important facet of cellular stress response. We recently demonstrated that this translational control involves a stress-specific reprogramming of modified ribonucleosides in tRNA. -

Protein Name Accession Number Molecular Weight Myovi-GTD
MyoVI-GTD MyoVI-GTD MyoVa-MGT MyoVa-MGT Molecular Spectral Unique Spectral Unique Protein Name Accession Number Weight Counts Peptides Counts Peptides Dync1h1 Cytoplasmic dynein 1 heavy chain 1 IPI00119876 532 kDa 310 121 515 182 Spna2 Spectrin alpha 2 IPI00757353 285 kDa 853 170 597 149 Myo5a 215 kDa protein IPI00875222 215 kDa 162 47 874 109 AU042671 hypothetical protein LOC269700 isoform 1 IPI00762814 453 kDa 2 2 231 104 Spnb2 Isoform 1 of Spectrin beta chain, brain 1 IPI00319830 274 kDa 505 122 347 100 Dmxl2 Isoform 1 of DmX-like protein 2 IPI00853932 338 kDa 63 38 251 100 Cltc Clathrin heavy chain 1 IPI00169916 (+1) 192 kDa 1994 138 565 90 Mtap2 12 days embryo spinal cord cDNA, RIKEN full-length enriched library, clone:C530026F16 product:microtubule-associated protein 2, full insert sequenceIPI00894724 199 kDa 229 82 258 74 Mtap1a Isoform 1 of Microtubule-associated protein 1A IPI00408909 (+1) 300 kDa 310 86 214 74 Itpr1 Isoform 4 of Inositol 1,4,5-trisphosphate receptor type 1 IPI00230019 (+3) 311 kDa 37 18 155 73 Huwe1 HECT, UBA and WWE domain containing 1 IPI00463909 (+1) 483 kDa 5 5 91 69 Fasn Fatty acid synthase IPI00113223 272 kDa 24 17 140 68 Usp9x Ubiquitin carboxyl-terminal hydrolase IPI00798468 291 kDa 68 45 98 65 Lrp1 Prolow-density lipoprotein receptor-related protein 1 precursor IPI00119063 505 kDa 92 53 109 62 Myh10 Myosin-10 IPI00515398 (+1) 229 kDa 65 40 98 59 Mical1 NEDD9-interacting protein with calponin homology and LIM domains IPI00116371 117 kDa 2 2 203 57 Plec1 Isoform PLEC-1I of Plectin-1 IPI00229509 (+10) -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -
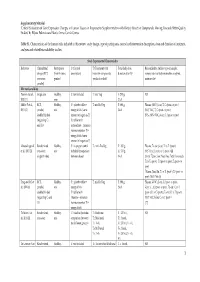
Critical Evaluation of Gene Expression Changes in Human Tissues In
Supplementary Material ‘Critical Evaluation of Gene Expression Changes in Human Tissues in Response to Supplementation with Dietary Bioactive Compounds: Moving Towards Better-Quality Studies’ by Biljana Pokimica and María-Teresa García-Conesa Table S1. Characteristics of the human trials included in this review: study design, type of participants, control and intervention description, dose and duration of treatment, analyses and related bioavailability studies. Study Experimental Characteristics Reference Clinical trial Participants C (Control T (Treatment with Total daily dose, Bioavailability studies: type of sample, design (RCT, (health status, description) bioactive compounds, duration (d or h)1 compounds and (or) metabolites analysed, crossover, gender) products or diet) main results2 parallel) Mix meals and diets Persson I et al., Single arm Healthy, C: not included T: mix Veg T: 250 g, NR 2000 [1] men 21 d Møller P et al., RCT, Healthy, C1: placebo tablet + T: mix FruVeg T: 600 g, Plasma: (NS↑) β-car, T, C2 (post- vs pre-) 2003 [2] parallel, mix energy drink (same 24 d (NC) VitC, T, C2 (post- vs pre-) double blinded amount of sugars as T) (NS↓, 69%) VitC, β-car, C1 (post- vs pre-) (regarding C1 C2: tablet with and C2) antioxidants + minerals (same amount as T) + energy drink (same amount of sugars as T) Almendingen K Randomized, Healthy, C: no proper control T1,2: mix FruVeg T1: 300 g, Plasma: ↑α-car, β-car, T2 vs T1 (post-) et al., 2005 [3] crossover, mix included (comparison T2: 750 g, (NS↑) Lyc, Lut, T2 vs T1 (post-) [4] single -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Structure of Human Aspartyl Aminopeptidase Complexed With
Chaikuad et al. BMC Structural Biology 2012, 12:14 http://www.biomedcentral.com/1472-6807/12/14 RESEARCH ARTICLE Open Access Structure of human aspartyl aminopeptidase complexed with substrate analogue: insight into catalytic mechanism, substrate specificity and M18 peptidase family Apirat Chaikuad1, Ewa S Pilka1, Antonio De Riso2, Frank von Delft1, Kathryn L Kavanagh1, Catherine Vénien-Bryan2, Udo Oppermann1,3 and Wyatt W Yue1* Abstract Backround: Aspartyl aminopeptidase (DNPEP), with specificity towards an acidic amino acid at the N-terminus, is the only mammalian member among the poorly understood M18 peptidases. DNPEP has implicated roles in protein and peptide metabolism, as well as the renin-angiotensin system in blood pressure regulation. Despite previous enzyme and substrate characterization, structural details of DNPEP regarding ligand recognition and catalytic mechanism remain to be delineated. Results: The crystal structure of human DNPEP complexed with zinc and a substrate analogue aspartate-β- hydroxamate reveals a dodecameric machinery built by domain-swapped dimers, in agreement with electron microscopy data. A structural comparison with bacterial homologues identifies unifying catalytic features among the poorly understood M18 enzymes. The bound ligands in the active site also reveal the coordination mode of the binuclear zinc centre and a substrate specificity pocket for acidic amino acids. Conclusions: The DNPEP structure provides a molecular framework to understand its catalysis that is mediated by active site loop swapping, a mechanism likely adopted in other M18 and M42 metallopeptidases that form dodecameric complexes as a self-compartmentalization strategy. Small differences in the substrate binding pocket such as shape and positive charges, the latter conferred by a basic lysine residue, further provide the key to distinguishing substrate preference. -

Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle
Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, Mekki Boussaha, Fabio Pilla, Baldassare Portolano, Salvatore Mastrangelo To cite this version: Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, et al.. Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle. Frontiers in Genetics, Frontiers, 2021, 10.3389/fgene.2021.675569. hal-03210766 HAL Id: hal-03210766 https://hal.inrae.fr/hal-03210766 Submitted on 28 Apr 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ORIGINAL RESEARCH published: 28 April 2021 doi: 10.3389/fgene.2021.675569 Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa 1, Gabriele Senczuk 2, Elena Ciani 3, Roberta -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated.