Free Energy. Thermodynamic Identities. Phase Transitions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamic Potentials and Natural Variables
Revista Brasileira de Ensino de Física, vol. 42, e20190127 (2020) Articles www.scielo.br/rbef cb DOI: http://dx.doi.org/10.1590/1806-9126-RBEF-2019-0127 Licença Creative Commons Thermodynamic Potentials and Natural Variables M. Amaku1,2, F. A. B. Coutinho*1, L. N. Oliveira3 1Universidade de São Paulo, Faculdade de Medicina, São Paulo, SP, Brasil 2Universidade de São Paulo, Faculdade de Medicina Veterinária e Zootecnia, São Paulo, SP, Brasil 3Universidade de São Paulo, Instituto de Física de São Carlos, São Carlos, SP, Brasil Received on May 30, 2019. Revised on September 13, 2018. Accepted on October 4, 2019. Most books on Thermodynamics explain what thermodynamic potentials are and how conveniently they describe the properties of physical systems. Certain books add that, to be useful, the thermodynamic potentials must be expressed in their “natural variables”. Here we show that, given a set of physical variables, an appropriate thermodynamic potential can always be defined, which contains all the thermodynamic information about the system. We adopt various perspectives to discuss this point, which to the best of our knowledge has not been clearly presented in the literature. Keywords: Thermodynamic Potentials, natural variables, Legendre transforms. 1. Introduction same statement cannot be applied to the temperature. In real fluids, even in simple ones, the proportionality Basic concepts are most easily understood when we dis- to T is washed out, and the Internal Energy is more cuss simple systems. Consider an ideal gas in a cylinder. conveniently expressed as a function of the entropy and The cylinder is closed, its walls are conducting, and a volume: U = U(S, V ). -
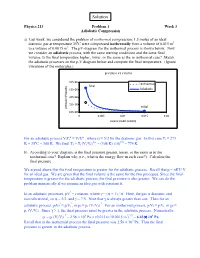
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

PDF Version of Helmholtz Free Energy
Free energy Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS which leads to dU - TdS ≤ 0 Since T and V are constant we can write this as d(U - TS) ≤ 0 The quantity in parentheses is a measure of the spontaneity of the system that depends on known state functions. Definition of Helmholtz Free Energy We define a new state function: A = U -TS such that dA ≤ 0. We call A the Helmholtz free energy. At constant T and V the Helmholtz free energy will decrease until all possible spontaneous processes have occurred. At that point the system will be in equilibrium. The condition for equilibrium is dA = 0. A time Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. The condition for spontaneous change is that ∆A is less than zero and the condition for equilibrium is that ∆A = 0. We write ∆A = ∆U – T∆S ≤ 0 (at constant T and V) Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. -

Lecture 4: 09.16.05 Temperature, Heat, and Entropy
3.012 Fundamentals of Materials Science Fall 2005 Lecture 4: 09.16.05 Temperature, heat, and entropy Today: LAST TIME .........................................................................................................................................................................................2� State functions ..............................................................................................................................................................................2� Path dependent variables: heat and work..................................................................................................................................2� DEFINING TEMPERATURE ...................................................................................................................................................................4� The zeroth law of thermodynamics .............................................................................................................................................4� The absolute temperature scale ..................................................................................................................................................5� CONSEQUENCES OF THE RELATION BETWEEN TEMPERATURE, HEAT, AND ENTROPY: HEAT CAPACITY .......................................6� The difference between heat and temperature ...........................................................................................................................6� Defining heat capacity.................................................................................................................................................................6� -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Thermodynamics, Flame Temperature and Equilibrium
Thermodynamics, Flame Temperature and Equilibrium Combustion Summer School 2018 Prof. Dr.-Ing. Heinz Pitsch Course Overview Part I: Fundamentals and Laminar Flames • Introduction • Fundamentals and mass balances of combustion systems • Thermodynamic quantities • Thermodynamics, flame • Flame temperature at complete temperature, and equilibrium conversion • Governing equations • Chemical equilibrium • Laminar premixed flames: Kinematics and burning velocity • Laminar premixed flames: Flame structure • Laminar diffusion flames • FlameMaster flame calculator 2 Thermodynamic Quantities First law of thermodynamics - balance between different forms of energy • Change of specific internal energy: du specific work due to volumetric changes: δw = -pdv , v=1/ρ specific heat transfer from the surroundings: δq • Related quantities specific enthalpy (general definition): specific enthalpy for an ideal gas: • Energy balance for a closed system: 3 Multicomponent system • Specific internal energy and specific enthalpy of mixtures • Relation between internal energy and enthalpy of single species 4 Multicomponent system • Ideal gas u and h only function of temperature • If cpi is specific heat at constant pressure and hi,ref is reference enthalpy at reference temperature Tref , temperature dependence of partial specific enthalpy is given by • Reference temperature may be arbitrarily chosen, most frequently used: Tref = 0 K or Tref = 298.15 K 5 Multicomponent system • Partial molar enthalpy hi,m is and its temperature dependence is where the molar specific -

Work and Energy Summary Sheet Chapter 6
Work and Energy Summary Sheet Chapter 6 Work: work is done when a force is applied to a mass through a displacement or W=Fd. The force and the displacement must be parallel to one another in order for work to be done. F (N) W =(Fcosθ)d F If the force is not parallel to The area of a force vs. the displacement, then the displacement graph + W component of the force that represents the work θ d (m) is parallel must be found. done by the varying - W d force. Signs and Units for Work Work is a scalar but it can be positive or negative. Units of Work F d W = + (Ex: pitcher throwing ball) 1 N•m = 1 J (Joule) F d W = - (Ex. catcher catching ball) Note: N = kg m/s2 • Work – Energy Principle Hooke’s Law x The work done on an object is equal to its change F = kx in kinetic energy. F F is the applied force. 2 2 x W = ΔEk = ½ mvf – ½ mvi x is the change in length. k is the spring constant. F Energy Defined Units Energy is the ability to do work. Same as work: 1 N•m = 1 J (Joule) Kinetic Energy Potential Energy Potential energy is stored energy due to a system’s shape, position, or Kinetic energy is the energy of state. motion. If a mass has velocity, Gravitational PE Elastic (Spring) PE then it has KE 2 Mass with height Stretch/compress elastic material Ek = ½ mv 2 EG = mgh EE = ½ kx To measure the change in KE Change in E use: G Change in ES 2 2 2 2 ΔEk = ½ mvf – ½ mvi ΔEG = mghf – mghi ΔEE = ½ kxf – ½ kxi Conservation of Energy “The total energy is neither increased nor decreased in any process. -

( ∂U ∂T ) = ∂CV ∂V = 0, Which Shows That CV Is Independent of V . 4
so we have ∂ ∂U ∂ ∂U ∂C =0 = = V =0, ∂T ∂V ⇒ ∂V ∂T ∂V which shows that CV is independent of V . 4. Using Maxwell’s relations. Show that (∂H/∂p) = V T (∂V/∂T ) . T − p Start with dH = TdS+ Vdp. Now divide by dp, holding T constant: dH ∂H ∂S [at constant T ]= = T + V. dp ∂p ∂p T T Use the Maxwell relation (Table 9.1 of the text), ∂S ∂V = ∂p − ∂T T p to get the result ∂H ∂V = T + V. ∂p − ∂T T p 97 5. Pressure dependence of the heat capacity. (a) Show that, in general, for quasi-static processes, ∂C ∂2V p = T . ∂p − ∂T2 T p (b) Based on (a), show that (∂Cp/∂p)T = 0 for an ideal gas. (a) Begin with the definition of the heat capacity, δq dS C = = T , p dT dT for a quasi-static process. Take the derivative: ∂C ∂2S ∂2S p = T = T (1) ∂p ∂p∂T ∂T∂p T since S is a state function. Substitute the Maxwell relation ∂S ∂V = ∂p − ∂T T p into Equation (1) to get ∂C ∂2V p = T . ∂p − ∂T2 T p (b) For an ideal gas, V (T )=NkT/p,so ∂V Nk = , ∂T p p ∂2V =0, ∂T2 p and therefore, from part (a), ∂C p =0. ∂p T 98 (a) dU = TdS+ PdV + μdN + Fdx, dG = SdT + VdP+ Fdx, − ∂G F = , ∂x T,P 1 2 G(x)= (aT + b)xdx= 2 (aT + b)x . ∂S ∂F (b) = , ∂x − ∂T T,P x,P ∂S ∂F (c) = = ax, ∂x − ∂T − T,P x,P S(x)= ax dx = 1 ax2. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

7 Apr 2021 Thermodynamic Response Functions . L03–1 Review Of
7 apr 2021 thermodynamic response functions . L03{1 Review of Thermodynamics. 3: Second-Order Quantities and Relationships Maxwell Relations • Idea: Each thermodynamic potential gives rise to several identities among its second derivatives, known as Maxwell relations, which express the integrability of the fundamental identity of thermodynamics for that potential, or equivalently the fact that the potential really is a thermodynamical state function. • Example: From the fundamental identity of thermodynamics written in terms of the Helmholtz free energy, dF = −S dT − p dV + :::, the fact that dF really is the differential of a state function implies that @ @F @ @F @S @p = ; or = : @V @T @T @V @V T;N @T V;N • Other Maxwell relations: From the same identity, if F = F (T; V; N) we get two more relations. Other potentials and/or pairs of variables can be used to obtain additional relations. For example, from the Gibbs free energy and the identity dG = −S dT + V dp + µ dN, we get three relations, including @ @G @ @G @S @V = ; or = − : @p @T @T @p @p T;N @T p;N • Applications: Some measurable quantities, response functions such as heat capacities and compressibilities, are second-order thermodynamical quantities (i.e., their definitions contain derivatives up to second order of thermodynamic potentials), and the Maxwell relations provide useful equations among them. Heat Capacities • Definitions: A heat capacity is a response function expressing how much a system's temperature changes when heat is transferred to it, or equivalently how much δQ is needed to obtain a given dT . The general definition is C = δQ=dT , where for any reversible transformation δQ = T dS, but the value of this quantity depends on the details of the transformation. -

Muddiest Point – Entropy and Reversible I Am Confused About Entropy and How It Is Different in a Reversible Versus Irreversible Case
1 Muddiest Point { Entropy and Reversible I am confused about entropy and how it is different in a reversible versus irreversible case. Note: Some of the discussion below follows from the previous muddiest points comment on the general idea of a reversible and an irreversible process. You may wish to have a look at that comment before reading this one. Let's talk about entropy first, and then we will consider how \reversible" gets involved. Generally we divide the universe into two parts, a system (what we are studying) and the surrounding (everything else). In the end the total change in the entropy will be the sum of the change in both, dStotal = dSsystem + dSsurrounding: This total change of entropy has only two possibilities: Either there is no spontaneous change (equilibrium) and dStotal = 0, or there is a spontaneous change because we are not at equilibrium, and dStotal > 0. Of course the entropy change of each piece, system or surroundings, can be positive or negative. However, the second law says the sum must be zero or positive. Let's start by thinking about the entropy change in the system and then we will add the entropy change in the surroundings. Entropy change in the system: When you consider the change in entropy for a process you should first consider whether or not you are looking at an isolated system. Start with an isolated system. An isolated system is not able to exchange energy with anything else (the surroundings) via heat or work. Think of surrounding the system with a perfect, rigid insulating blanket. -

Chapter 3 3.4-2 the Compressibility Factor Equation of State
Chapter 3 3.4-2 The Compressibility Factor Equation of State The dimensionless compressibility factor, Z, for a gaseous species is defined as the ratio pv Z = (3.4-1) RT If the gas behaves ideally Z = 1. The extent to which Z differs from 1 is a measure of the extent to which the gas is behaving nonideally. The compressibility can be determined from experimental data where Z is plotted versus a dimensionless reduced pressure pR and reduced temperature TR, defined as pR = p/pc and TR = T/Tc In these expressions, pc and Tc denote the critical pressure and temperature, respectively. A generalized compressibility chart of the form Z = f(pR, TR) is shown in Figure 3.4-1 for 10 different gases. The solid lines represent the best curves fitted to the data. Figure 3.4-1 Generalized compressibility chart for various gases10. It can be seen from Figure 3.4-1 that the value of Z tends to unity for all temperatures as pressure approach zero and Z also approaches unity for all pressure at very high temperature. If the p, v, and T data are available in table format or computer software then you should not use the generalized compressibility chart to evaluate p, v, and T since using Z is just another approximation to the real data. 10 Moran, M. J. and Shapiro H. N., Fundamentals of Engineering Thermodynamics, Wiley, 2008, pg. 112 3-19 Example 3.4-2 ---------------------------------------------------------------------------------- A closed, rigid tank filled with water vapor, initially at 20 MPa, 520oC, is cooled until its temperature reaches 400oC.