Spectroscopic and Theoretical Investigations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Essays in Public Economics by Dorian Carloni a Dissertation Submitted In
Essays in Public Economics by Dorian Carloni A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Economics in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Alan J. Auerbach, Chair Professor Hilary W. Hoynes Professor Rucker C. Johnson Professor Emmanuel Saez Spring 2016 Essays in Public Economics Copyright 2016 by Dorian Carloni 1 Abstract Essays in Public Economics by Dorian Carloni Doctor of Philosophy in Economics University of California, Berkeley Professor Alan J. Auerbach, Chair This dissertation consists of three chapters and analyzes individuals' and firms’ response to tax and government spending policies. The first chapter focuses on the economic incidence of a large value added tax (VAT) cut in the restaurant industry in France. In particular it estimates the share of the tax cut falling on workers, firm owners and consumers by analyzing the VAT cut applied to French sit-down restaurants { a drop from 19.6 percent to 5.5 percent { in July 2009. Theoretically, we develop a standard partial equilibrium model of consumption tax incidence that includes consumption substitutability between the taxed good and an untaxed good, and markets for production inputs, which we allow to vary with the tax. Empirically, we use firm-level data and a difference-in-differences strategy to show that the reform increased restaurant profits and the cost of employees, and aggregate price data to estimate the decrease in prices produced by the reform. We compare sit-down restaurants to (a) non-restaurant market services and (b) non-restaurant small firms, and find that prices decreased by around 2 percent, the cost per employee increased by 3.9 percent and the return to capital increased by around 10 percent. -

Ocn752505145-2018-11-02-By-Last-Name.Pdf (248.0Kb)
System Inspectors Sorted by Last Name Total Count: 1424 SI# Expires Name Association Town State 14215 5/1/2021 Zachary D. Aaronson CDM Smith Providence RI 4698 6/30/2019 James Abare Winchendon MA 4046 6/30/2019 Stacey J. Abato Raggs, Inc. Concord MA 2548 6/30/2019 Carl Adamo Lincoln RI 349 6/30/2019 Stephen M. Adams Norwell MA 13724 6/30/2019 David R. Adler Charlton MA 14072 12/1/2019 Nicholas Aguiar Yarmouth DPW - Engineering Division West Yarmouth MA 4332 6/30/2019 James D. Aguiar, Jr. Tri-Spec Corp. Westport MA 14016 5/1/2020 Thorsen Akerley Williams & Sparages, LLC Middleton MA 13661 6/30/2019 Patrick Alderman Swansea MA 2040 6/30/2019 Michael Alesse East Coast Excavating Groveland MA 4789 6/30/2019 Lisa C. Allain Millbury MA 4955 6/30/2019 Mark Allain Peabody MA 12864 6/30/2020 William Allen Bill Allen Enterprises LLC Spring Hill FL 13850 6/30/2021 Edward W. Alleva Jr. Alleva Excavating Company Lunenburg MA 350 6/30/2019 Karl J. Alm Alm and Son Septic West Brookfield MA 14169 12/1/2020 Wyatt J. Alm West Brookfield MA 13975 4/1/2019 Elizabeth Alves All Clear Title 5 Services Norton MA 14252 8/1/2021 Nathon Amaral Mystic Excavating Corp Raynham MA 13218 6/30/2021 Erik Anderson Norwell MA 4658 6/30/2019 Michael Andrade Graves Engineering, Inc. Sutton MA 14210 5/1/2021 Samantha Andrews T. Silvia Excavating Swansea MA 2666 6/30/2019 John R. Andrews, III Andrews Survey & Engineers, Inc. -

Entering New Waters at Pier 94 Leslie Weir
the newsletter of the golden gate audubon society // vol. 103 no. 4 fall 2019 EntEring nEw watErs at PiEr 94 leslie weir ore than a million individual shore- M birds rely on the San Francisco Bay for at least some portion of their annual lifecycle. The Bay Area is classified as a site of hemispheric significance for migra- tory shorebirds by the Western Hemisphere Shorebird Reserve Network, and the Ramsar Convention on Wetlands has designated San Francisco Bay as a site of international sig- nificance for migratory waterfowl. More than 1,000 species of birds, mammals, and fish rely on the San Francisco Bay Estuary—the centerpiece of our region. CONTINUED on page 5 American Avocet at Pier 94. Bob Gunderson long enough to be restored for his aston- ishing journey. Ornithologists discovered that Blackpoll Warblers have the longest migratory route of any New World warbler. Every fall, these tiny 12-gram birds make a nonstop transatlantic flight from upper Canada to South America. Then, the fol- lowing spring, they reverse migrate to their breeding grounds. A few hours later, I checked on this exquisite feathered jewel. He was no longer quiet. I could hear him bouncing against the box top in a frenzied urge to leap up and fly south. I let him go and off he went—toward the Statue of Liberty, a fitting testament to his fortitude. I hoped that I had helped him a little. Cindy Margulis Like this Blackpoll Warbler’s drive to Male Blackpoll Warbler. migrate during the fall season, this is our time of transition at GGAS. -

Priests and Deacons INDEX Archdiocesan Priests
Priests and Deacons INDEX Archdiocesan Priests ..................................................................................................... 1 Priests of the Archdiocese in the Order of Ordination ................................................. 14 Priests and Deacons: Alphabetical Necrology ............................................................ 21 Priests and Deacons: Chronological Necrology by Month, Day and Year .................. 51 Archdiocesan Permanent Deacons ............................................................................ 79 11/30/2020 Archdiocesan Priests Adams, Rev. John E. (1969) Baer, Rev. Timothy K. (1996) Director, S.O.M.E., DC Pastor, St. Nicholas, Laurel Phone: 202-832-9710 Phone: 301-490-5116 Aguirre, Rev. Francisco E. (2013) Baraki, Rev. Tesfamariam (1975) Pastor, St. Catherine Labouré, Chaplain, Howard University Hospital, Wheaton DC Phone: 301-946-3636 In Residence, St. Gabriel, DC Phone: 202-726-9092 Agustin Rev. Patrick S. (2020) Parochial Vicar (pro-tem), St. Francis of Barbieri de Carvalho, Rev. Rafael Assisi, Dearwood (2013) Phone: 301-840-1407 Director of Spiritual Formation, Redemptoris Mater Archdiocesan Ailer, Rev. Gellert J. (2006) Missionary Seminary, Hyattsville Missionary Assignment, Archdiocese of Eger, Hungary Barry, Rev. John M. (1988) Pastor, Church of the Resurrection, Alliata, Rev. Peter R. (1961) Burtonsville Retired, St. John Vianney, Prince Phone: 301-236-5200 Frederick Phone: 301-440-1784 Bava, Rev. David A. (1973) Pastor, Holy Redeemer, DC Amey, Rev. Msgr. Robert -
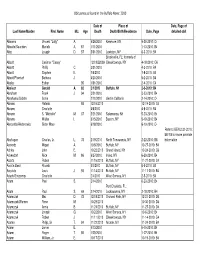
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

Quarta-Feira, 15 De Maio De 2013
iário ficial Nº 10.627 - Ano XLIIID OPrefeitura Municipal de Campinas Quarta-feira, 15 de maio de 2013 www.campinas.sp.gov.br MÉRCIO E SAÚDE ANIMAL LTDA. EPP em relação à especifi cação do item 1 GABINETE DO PREFEITO descrita no Anexo I - Especifi cações dos Itens do edital, conforme segue: 1. A respeito das exigências do item 01, ração para cães fi lhotes, acreditamos que os níveis de garantia do alimento para cães fi lhotes devam ser maiores que os níveis DECRETO Nº 17.970 DE 14 DE MAIO DE 2013 de garantia do alimento para cães adultos. Normalmente o nível de proteína é de, no AUTORIZA DESCONTO AOS PROFESSORES E ESTUDANTES DAS RE- mínimo, 30% devido o animal em crescimento necessitar de mais nutrientes do que DES MUNICIPAL E ESTADUAL DE ENSINO NO PREÇO DO INGRES- um animal adulto. SO DOS CONCERTOS DA ORQUESTRA SINFÔNICA MUNICIPAL DE Resposta: Será disponibilizado novo Edital prevendo ração para cães fi lhotes com CAMPINAS E DÁ OUTRAS PROVIDÊNCIAS. PROTEÍNA BRUTA min. 32% e máx. 35%. O Prefeito do Município de Campinas, no uso de suas atribuições legais, Campinas, 14 de maio de 2013 DECRETA : JOAO FERNANDES FILHO Art. 1° Fica autorizado desconto aos professores e estudantes das Redes Municipal e Pregoeiro Estadual de Ensino no preço do ingresso dos concertos da Orquestra Sinfônica Mu- nicipal de Campinas que se realizarem nas dependências dos teatros e auditórios mu- EXPEDIENTE DESPACHADO PELO SR.SECRETÁRIO nicipais. MUNICIPAL DE ADMINISTRAÇÃO Art. 2º Para atendimento do disposto no art. 1º serão pagos os seguintes valores, por DECLARAÇÃO DE ITEM FRACASSADO E HOMOLOGAÇÃO ingresso: Processo Administrativo n º 12/10/56.360 I - 1/3 (um terço) do valor do ingresso para os professores; Interessado: Secretaria Municipal de Saúde II - 1/6 (um sexto) do valor do ingresso para os alunos. -

Not One Bomb for Iran Petition We the People of the United States Of
Not One Bomb For Iran Petition We the people of the United States of America are deeply concerned about the impending threat a nuclear capable Iran poses for both Israel and the United States. We believe that America and its allies must demand that Iran fully dismantle its nuclear program. We do not believe Iran can be trusted to simply keep its word. Consequently, we believe any agreement between the United States and Iran must conform to the following two standards: 1. Any agreement the Administration signs with Iran must completely dismantle the regime’s ability to build a nuclear bomb. 2. Congress must ratify any agreement proposed by the Administration with regard to Iran’s nuclear program. As the people of the United States we must settle for nothing less. Sincerely, 1 Kirstie Ansell 2 Darrell Law 3 Andrew Gregory 4 Mark Dreistadt 5 Joseph Coram 6 Randall Fahey 7 Fred Parkhill 8 Dale Ulmer 9 Loretta Bodtmann 10 Richard Eisenberg 11 Gila Grossman 12 Joyce Backman 13 Margaret Wagner 14 John Sherwood 15 Dolores Coronel 16 Lynda Higginbotham 17 Arleta Stewart 18 Elsie Johnson 19 Vickie Boyd 20 Audrey Johnsen 21 Joann Willer 22 Carol Neagles 23 Connie Mclain 24 Jean Johnston 25 Laura Martin 26 Patricia Christner 27 Mercile Martinsen 28 Sadie Slack 29 Glenda Morris 30 Mary Elerding 31 Cheryllynn Folk 32 William T Lewis 33 Linda Holbert 34 Jerry Duckworth 35 Lawana Rasmussen 36 Jerry Duckworth 37 Ron Blankenship 38 T Simons 39 Chris Lee 40 Dale Kimberlin 41 Curtis Guest 42 Meredith Moreland 43 Maureen Hart 44 Anne Barnhart 45 James Johnson -

Aspects of Gravitational Collapse and the Formation of Spacetime Singularities
Aspects of Gravitational Collapse and the formation of Spacetime Singularities Soumya Chakrabarti Department of Physical Sciences Indian Institute of Science Education and Research Kolkata Supervisor: Prof. Narayan Banerjee arXiv:1709.01512v1 [gr-qc] 5 Sep 2017 Thesis submitted to IISER Kolkata for the fulfilment of the requirements for the Degree of Doctor of Philosophy September 2017 To Mother... Declaration I certify that this work contains no material which has been accepted for the award of any other degree or diploma in my name, in any university or other tertiary institution, to the best of my knowledge and belief, contains no material previously published or written by another person, except where due reference has been made in the text and acknowledgement of collaborative research. In addition, I certify that no part of this work will, in the future, be used in a submission in my name, for any other degree or diploma in any university or other tertiary institution without the prior approval of Indian Institute of Science Education and Research Kolkata, India and collaborators. Soumya Chakrabarti Registration number: 10IP11 Department of Physical Sciences IISER Kolkata September 2017 Certificate This is to certify that the Ph.D. thesis entitled "Aspects of Gravitational Collapse and the formation of Spacetime Singularities" submitted by Soumya Chakrabarti is absolutely based upon his own work under the supervision of Prof. Narayan Banerjee at the Indian Institute of Science Education and Research, Kolkata (IISER Kolkata) and that neither this thesis nor any part of it has been submitted for either any degree/diploma or any other academic award anywhere before. -

Sacred Heart of Jesus Parish Legacy of Faith Campaign Donations from Estates, in Honor Of, and in Memory Of
Sacred Heart of Jesus Parish Legacy of Faith Campaign Donations from Estates, in Honor of, and in Memory of FROM THE ESTATE OF Amelia Calcagni Lucy Tarca Claire Tosches IN HONOR OF DONOR Ann & Pat's 50th wedding anniversary Gift from Mr. & Mrs. Silvio Sannicandro Anthony F. Mussulli, Sr.'s 100th birthday celebration Gift from Stephen Minichiello Carl & Helen DiGregorio's 50th wedding anniversary Gift from Joe & Mary Arcudi Don & Jean Dauphinee's 53rd wedding anniversary Gift from the Bonina family Larry & Regina Del Vecchio Gift from Mr. & Mrs. Stephen Del Vecchio Larry & Regina Del Vecchio Gift from Mr. & Mrs. Stephen Del Vecchio Janice & Louis Guerriere's Golden Anniversary Gift from June Glennon Regina Mainini Gift from Julie Lobisser Reno & Jean DeLuzio's 50th Anniversary Gift from Dorothy & Dick Pedroli IN MEMORY OF DONOR Al, Curtis and Alex Gift from Elena Fontana Frank Andreotti Gift from John & Joanne Fortunato Louis Arcudi, Jr. Gift from Eleanor Boni Louis Arcudi, Jr. Gift from Ronald Abraham Louis Arcudi, Jr. Gift from Terri Moschilli Richard Barry Gift from Mary Lou Iacovelli Richard Barry Gift from Terri Moschilli Josephine Basile Gift from Grace Nealon Maria Beccia Gift from Charlene Antonellis Maria Beccia Gift from Charlie & Joan Pederzoli Paul Beccia Gift from Mary Lou Iacovelli John Bianchi Gift from Deacon Pat Mussulli Rose Byron Gift from Deacon Pasquale Mussulli Josephine Calzolaio Gfit fromCarole Beccia Rose Caprini Gift from Robert & Norma Murphy Richard Carloni Gift from Peggy, Kathy, June & Paula Peter Castagnaro Gift from James Mohan F. Lawrence Catella Gift from Mr. & Mrs. Peter Pessotti Phyllis M. -

Guidelines for Monitoring Autophagy in Higher Eukaryotes
Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (2nd edition) Daniel J Klionsky,1,2,* Kotb Abdelmohsen, Akihisa Abe, Md. Joynal Abedin, Hagai Abeliovich, Abraham Acevedo Arozena, Hiroaki Adachi, Christopher M Adams, Peter D Adams, Khosrow Adeli, Peter J. Adhihetty, Sharon G Adler, Galila Agam, Rajesh Agarwal, Manish K Aghi, Maria Agnello, Patrizia Agostinis, Julio Aguirre-Ghiso, Slimane Ait-Si-Ali, Takahiko Akematsu, Emmanuel T Akporiaye, Mohamed Al-Rubeai, Guillermo M Albaiceta, Diego Albani, Matthew L Albert, Jesus Aldudo, Hana Algül, Mehrdad Alirezaei, Iraide Alloza, Alexandru Almasan, Emad S Alnemri, Covadonga Alonso, Dario C Altieri, Lydia Alvarez-Erviti, Sandro Alves, Giuseppina Amadoro, Atsuo Amano, Consuelo Amantini, Santiago Ambrosio, Amal O Amer, Mohamed Amessou, Angelika Amon, Frank A. Anania, Stig U Andersen, Usha P Andley, Catherine K Andreadi, Nathalie Andrieu-Abadie, Alberto Anel, David K Ann, Shailendra Anoopkumar-Dukie, Hiroshi Aoki, Nadezda Apostolova, Saveria Aquila, Katia Aquilano, Koichi Araki, Eli Arama, Jun Araya, Alexandre Arcaro, Esperanza Arias, Hirokazu Arimoto, Aileen R Ariosa, Thierry Arnould, Ivica Arsov, Valerie Askanas, Eric Asselin, Ryuichiro Atarashi, Julie D Atkin, Laura D Attardi, Patrick Auberger, Georg Auburger, Laure Aurelian, Riccardo Autelli, Laura Avagliano, Maria Laura Avantaggiati, Laura Avagliano, Limor Avrahami, Tiziana Bachetti, Jonathan M Backer, Dong-Hun Bae, Ok-Nam Bae, Soo Han Bae, Eric H Baehrecke, Seung-Hoon Baek, Stephen Baghdiguian, Agnieszka Bagniewska-Zadworna, -

Town of Franklin 2004 Annual Report
TOWN OF FRANKLIN 2004 ANNUAL REPORT 2 In Memoriam Donald E. P eir ce July 21,1933 - January 28, 2003 Franklin Public Schools Mary M Ristaino July 13, 1921 - February 9, 2003 Franklin Public Schools Mar y “Sheila” Bur ke September l2, 1937 - March 15, 2003 Cable Rerun Coordinator Giustino A. Socci March 23, 1926 - March 18, 2003 Board of Health Angelina C. Pizzi November 21, 1922 - April 12, 2003 H.S. Cafeteria Worker Ang elina Wood August 21, 1928 - June 23, 2003 Local Artist/Franklin Prints Various Committees Nor ma A. Ar ruda August 1, 1933 - July 8, 2003 Election Worker Ev el yn T. Supple September 23, 1927 - August 3, 2003 Election Worker - Municipal Employee Jer emiah M. Scaccia July 15, 1914 - August 14, 2003 Various Committees Ver non R. Ander son July 26,1916 - October 20, 2003 School Custodian John J . Br ennan February 17, 1926 - December 24, 2003 Various Committees On behalf of the Town of Franklin, we offer our sincere appreciation to all these people that have taken the time to serve their community. We are forever thankful. 3 TABLE OF CONTENTS Animal Control ..................................................................................................................................................................................... 90 Assessors, Board of ............................................................................................................................................................................ 151 Town Financial Summary.................................................................................................................................................... -

Active Mooring List
"Snapshot" Active Mooring Permits 4/1/2021 HrbrMstr HID Last Name First Name Business Name Permit# pbName Length Delude Bellamy BETTENCOURT ARTHUR 1330 SYMPATICO 20.0 Bellamy DIGGINS TODD 7385 unk 13.0 Bellamy FITZGERALD TREVOR 7488 No Name Assigned 25.0 Bellamy GOLDACKER ROY K 6634 No Name Assigned 17.0 Bellamy GOVE RHONDA 7989 SLIPPERY 25.0 Bellamy HEANEY JAMIE A DOVER POINT OYSTER COMPANY 7730 Pontoon/Float 24.0 Bellamy HEANEY JAMIE A DOVER POINT OYSTER COMPANY 7731 Unknown 22.0 Bellamy HUGGINS CLEO 7743 FUNDECK 20.0 Bellamy MONBOUQUETTE MARC O 7396 UNKNOWN 23.0 Bellamy O'MALEY BRIAN 7563 UNKNOWN 10.0 Bellamy PARKER ERIC GRAYSON 6010 DNA 26.0 Bellamy RONALD JAMES ANDREW 7830 Carolina Skiff 21.0 Bellamy SCASCITELLI JOSEPH R 7753 No Name Assigned 10.0 Delude Cocheco - - GREAT BAY ROWING CLUB 4801 No Name Assigned 22.0 Cocheco - - GREAT BAY ROWING CLUB 4630 No Name Assigned 16.0 Cocheco - - GREAT BAY ROWING CLUB 7333 STARCRAFT 14.0 Delude Little Bay - - NEWICK'S LOBSTER HOUSE 4605 No Name Assigned 39.0 Little Bay - - NEWICK'S LOBSTER HOUSE 174 No Name Assigned 35.0 Little Bay - - NEWICK'S LOBSTER HOUSE 1061 No Name Assigned 29.0 Little Bay BARRETT NANCY 4945 OLIVE 24.0 (qryPermitListWebsitewithBusinessnames) 1 "Snapshot" Active Mooring Permits 4/1/2021 HrbrMstr HID Last Name First Name Business Name Permit# pbName Length Delude Little Bay BARSTOW DIANTHA 7319 No Name Assigned 16.0 JONATHAN Little Bay BUNKER ROBERT 7758 FLOAT 16.0 Little Bay CARTNICK EDWARD 4120 No Name Assigned 25.0 Little Bay CASSADY SHELDON MARK 7653 No Name Assigned